Spotlight on the Clinical Evaluation Process
More Than Data Collection and Reporting
“Clinical Evaluation better serves a manufacturer, when the mindset of the process changes from ‘a one-off document to generate’, to an ongoing process to be maintained. This understanding positively serves the medical device by identifying required refinements to associated documents such as instructions for use, claims and post-market processes, but also forewarning the manufacturer to take pre-emptive action for any possible gaps in evidence, risk or otherwise.”

by Dr Sylvia Lui
Clinical Affairs Analyst, SciMed Consultancy Ltd
EXECUTIVE SUMMARY
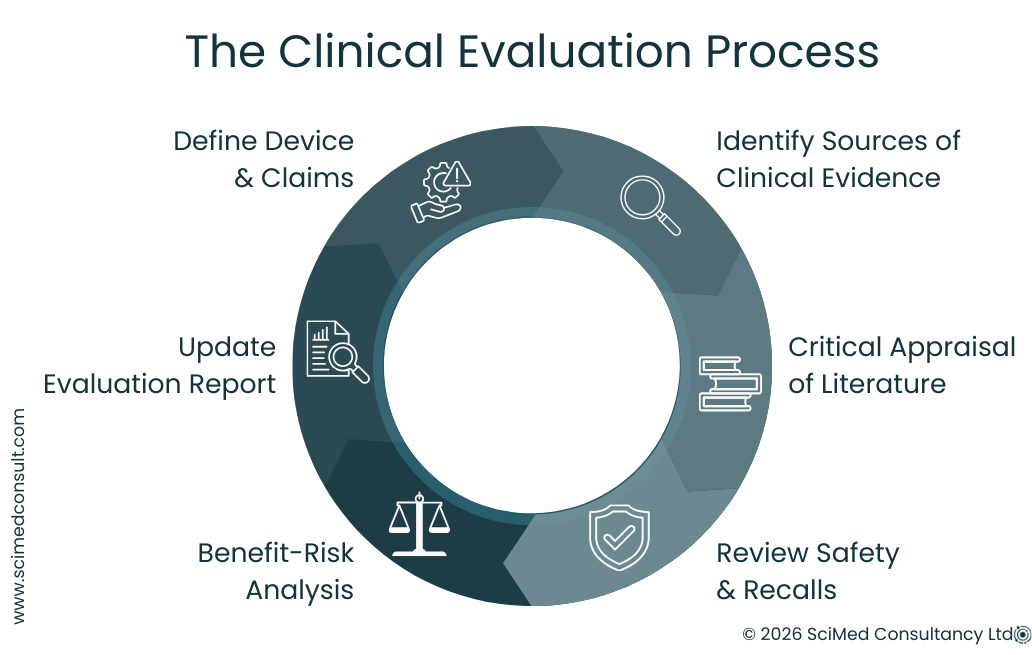
Clinical Evaluation is a process, not just a document. The reports seen by regulators reflect a broader analytical and decision-making workflow.
Structured planning is essential. Defining device purpose, claims, intended population and risk profile forms the foundation for meaningful evaluation.
Evidence must be critically appraised. Not all literature, safety notices or post-market data are relevant; inclusion requires careful judgement.
Interpretation matters. Numbers alone do not tell the story - recall data, safety notices, and literature findings require contextual analysis.
Benefit-risk is central. All evaluation activities support a transparent assessment of device performance, clinical benefit, and safety.
Clinical Evaluation is ongoing. The process should be actively maintained and updated throughout the device lifecycle.
INTRODUCTION
Clinical Evaluation often looks very different in practice from how it is commonly imagined. It is sometimes perceived as a process centred on form filling or routine reporting. While there are certainly spreadsheets, recurring reports and routine monitoring activities involved, these represent only a small part of the overall process.
In reality, Clinical Evaluation requires a significant amount of preparation, structured thinking and critical analysis. A central principle in modern regulatory frameworks is that Clinical Evaluation should function as a living process - one that is actively maintained, reviewed and updated throughout the lifecycle of a medical device.
The formal documentation seen in regulatory submissions is only the final output of this work. Behind each report is a series of decisions and analytical steps that determine whether a device can be clearly demonstrated to perform as intended, deliver clinical benefit and maintain an acceptable safety profile. Clinical Evaluation therefore involves judgement as well as documentation: deciding what evidence is relevant, what is not, and how that evidence supports conclusions about performance, safety and benefit-risk.
CLINICAL EVALUATION DOESN’T STAND STILL
Expectations around clinical evidence, lifecycle management, and audit readiness continue to evolve.
MedTech Horizon provides monthly guidance on clinical evaluation, CER development, and how regulatory teams are maintaining compliance in practice.
How It Begins
A significant proportion of Clinical Evaluation work begins with structured planning. Before evidence is gathered or analysed, it is necessary to define the device, identify existing sources of data and determine how that evidence will be located and assessed.
At its core, the objective is twofold:
To fully understand the device itself.
To understand the evidence that supports it.
Understanding the Device
Understanding the device begins with clearly defining its intended purpose, indications, target population, design characteristics and mode of action. Supporting information is typically found within technical documentation, instructions for use and internal design records.
Alongside this, a State of the Art assessment is required to establish the current clinical landscape. This review identifies existing clinical practices, comparable technologies and relevant standards. It provides the context necessary to interpret the device’s performance and safety within current medical knowledge.
Before evaluating evidence, it is also essential to clarify the device’s claims, expected clinical benefits, performance outcomes and safety endpoints. This includes identifying known and residual risks and considering how these risks are balanced against the anticipated benefits. A documented and traceable risk management process is therefore an important supporting component of Clinical Evaluation.
Establishing these elements creates a device-specific framework against which evidence can be assessed for relevance and quality. Without this structure, evidence searches can quickly become unfocused, generating large amounts of data that may not be meaningful for demonstrating safety or performance.
Understanding the Evidence
Once the device characteristics and evaluation methodology are defined, the next stage is identifying relevant sources of evidence. These may include:
Scientific and clinical literature
Regulatory publications and guidance
Safety notices and recall databases
Internal reports and post-market surveillance data
During literature searching and appraisal, several key questions guide the evaluation process:
Is this information relevant to the device, its indication or the intended user population?
Is the evidence of sufficient quality and reliability?
Does the finding represent an isolated observation or part of a broader trend?
Does the information introduce new risks or reinforce known ones?
Is it directly relevant to the device’s claims, safety profile or performance endpoints?
In practice, a large proportion of identified information does not pass these filters. Even peer-reviewed publications may appear relevant at first glance but ultimately prove clinically or technically unrelated to the device under evaluation.
Critical Appraisal and Analysis
An early lesson in Clinical Evaluation is that literature review is often less about summarising studies and more about determining which studies should not be included.
Published conclusions cannot simply be accepted at face value. Instead, evidence must be examined in the context of the specific device and its intended use. Many studies may appear technically related but have limited clinical relevance, while others may focus on similar clinical indications but involve different device technologies or endpoints.
Even well-designed studies with strong statistical methods may ultimately be of limited relevance if the device usage, patient population or clinical outcomes differ significantly from those being evaluated.
For this reason, literature review involves critical analysis and justified exclusion, not simply aggregation of published data. Developing the ability to determine what should be included - and what should not - is an important part of building experience in Clinical Evaluation.
Reviewing Your Clinical Evaluation Approach
Many teams complete Clinical Evaluation documentation, but few establish a process that remains consistent and defensible over time.
If you are reviewing how your approach holds up in practice, a structured assessment can help highlight where gaps may emerge.
»Access the Clinical Evaluation Audit Checklist
Safety and Recall Notices
Another commonly used evidence source is international safety and recall databases. When reviewing these data, several factors must be considered:
Where the device or device group is marketed
The number of recalls or safety notices reported over time
The severity and frequency of reported events
The relationship between reported events and the number of devices in use
The duration of time the device has been on the market
At first glance, some recall records may appear significant or unusually frequent. However, detailed examination often reveals that many notices relate to issues such as packaging errors, manufacturing variability or labelling corrections, which may not have direct implications for patient safety.
When reports do relate to patient impact, further analysis is required. This involves examining the cause of the report, its regulatory classification and the corrective actions taken. The aim is to determine whether these reports indicate an emerging safety trend relevant to the device under evaluation.
In this way, reviewing safety notices is not simply a matter of reporting numbers. It requires interpretation and contextual analysis.
Pro Tip
Clinical Evaluation is far easier to defend when it is built into the development process, rather than reconstructed later.
When planning is delayed, gaps in evidence, claims, and risk justification tend to surface during review — often when timelines are already under pressure.
IN CONCLUSION
Clinical Evaluation involves a wide range of preparatory and analytical activities that are not always visible in the final documentation. Much of this work is reflected in the preparation of State of the Art reviews, Clinical Evaluation Plans and Clinical Evaluation Reports.
Ultimately, Clinical Evaluation can be understood as a structured process built around clarity, analysis and judgement:
Clinical Evaluation is a process, not just a document.
It requires structured planning, evidence identification and critical analysis.
Evidence must be assessed for both relevance and quality.
Safety notices, recalls and literature findings require interpretation rather than simple reporting.
Data must be actively analysed to demonstrate safety, performance and benefit-risk.
The final report represents the outcome of these activities - the visible result of a much broader process of evidence assessment and regulatory justification.
If you are reviewing your clinical evaluation process or preparing for audit, you can speak with our team for a focused discussion.