MDR Clinical Data - New for 2026
What Counts, What Doesn’t and How to Stay Compliant
“Lifecycle management matters. The clinical evaluation process is ongoing; manufacturers must maintain a living CER and update PMCF activities as new evidence emerges.”

by Dr
Nadhim
Bayatti
Senior Analyst, SciMed Consultancy Ltd
NEW FOR 2026:
In December 2025, the European Commission published a proposal to simplify rules on medical and in vitro diagnostic devices. Released on 16 December 2025, by the Directorate-General for Health and Food Safety, it introduces targeted amendments aimed at reducing regulatory burden in EU MDR/IVDR (amending MDR 2017/745, IVDR 2017/746, EMA crisis framework 2022/123, and AI Act 2024/1689).
The proposal covers a wide range of topics and has implications for clinical evidence ‘modernisation’. This is what it means through the lens of clinical evidence requirements and the impact it may have on manufacturers.
A wider range of data may qualify as clinical data. The conditions for relying on clinical data of an equivalent device are made more flexible. In Article 61 MDR, the possibility to demonstrate a device’s safety and performance based on non-clinical data alone is expanded. The use of ‘New Approach Methodologies’, such as in silico testing, is promoted.
Equivalence reliance made more flexible: the requirement in Regulation (EU) 2017/745 for a contract with the manufacturer of the equivalent device granting access to its technical documentation should therefore be removed and the equivalence criteria be adapted.
“Well-established Technologies” definition introduced: a definition of ‘well-established technology device’ is introduced for devices which will be subject to more proportionate requirements, replacing the lists of devices in the current Articles 18(3), 52(4) and 61(6)(b) MDR. This is particularly relevant to legacy devices.
Source
Directorate-General for Health and Food Safety. Proposal for a regulation to simplify rules on medical and in vitro diagnostic devices - Public Health (2025).
EXECUTIVE SUMMARY
The MDR has significantly raised the bar for what qualifies as acceptable clinical data. Evidence that was sufficient under the MDD - such as literature reviews or indirect equivalence claims - may no longer meet current data requirements.
Manufacturers now face tighter scrutiny from Notified Bodies. Inadequate or poorly justified clinical evidence is a leading cause of MDR certification delays, additional study requirements, and even market withdrawals.
A structured, device-specific approach to the clinical evaluation process is essential. Early development of a CEP, transparent justification of clinical data sources, and proactive PMCF activities are now fundamental to demonstrating MDR compliance.
Legacy devices transitioning from MDD to MDR must re-evaluate their clinical evidence base. Reliance on lower-tier or cumulative data sources must be supported by clear scientific justification aligned with MDCG 2020-6 guidance.
An integrated, lifecycle-based evidence strategy - combining clinical evaluation, PMCF, and continuous data updates - provides the most effective path to sustaining MDR compliance and market access.
INTRODUCTION
Since its introduction in 2021, the European Medical Device Regulation (MDR) has increased requirements for the scope and quality of clinical evidence needed to demonstrate device safety and performance. What once sufficed under the Medical Devices Directive (MDD) - such as literature reviews or equivalence claims - is no longer automatically acceptable.
For manufacturers and regulatory teams, understanding what counts as acceptable clinical data under the MDR - and what doesn’t - is now essential to maintain certification, avoid delays, and ensure continued market access. The MDR has also made clinical evaluation a continuous, lifecycle-based process, requiring ongoing Post-Market Clinical Follow-Up (PMCF) and updates to clinical evidence.
This article outlines the types of clinical data sources that are accepted, identifies common sources of non-compliance, and provides practical guidance for building a robust, compliant clinical evidence strategy under the MDR framework.
THE MDR: RAISING THE BAR ON CLINICAL DATA
Under the previous MDD, manufacturers had more flexibility in demonstrating safety and performance. Many legacy devices relied on equivalence claims or literature reviews without direct clinical investigations.
The MDR Article 61 has significantly raised expectations. It explicitly requires that clinical evaluations rely on ‘sufficient clinical evidence’ derived from acceptable data sources. This evidence must:
Confirm compliance with the General Safety and Performance Requirements (GSPRs),
Assess potential side effects,
Demonstrate a favourable benefit-risk ratio.
Additionally, MDR mandates continuous updates to clinical data throughout the device lifecycle, with obligatory PMCF to monitor real-world safety and performance.
Key Implications for Manufacturers
Device-specific data is now critical. Generic literature or equivalence claims are no longer sufficient without rigorous justification.
Notified Bodies are scrutinizing data quality and structure. Vague claims, incomplete datasets, or poorly justified equivalence are common reasons for submission delays or rejection.
Lifecycle management matters. The clinical evaluation process is ongoing; manufacturers must maintain a living Clinical Evaluation Report (CER) and update PMCF activities as new evidence emerges.
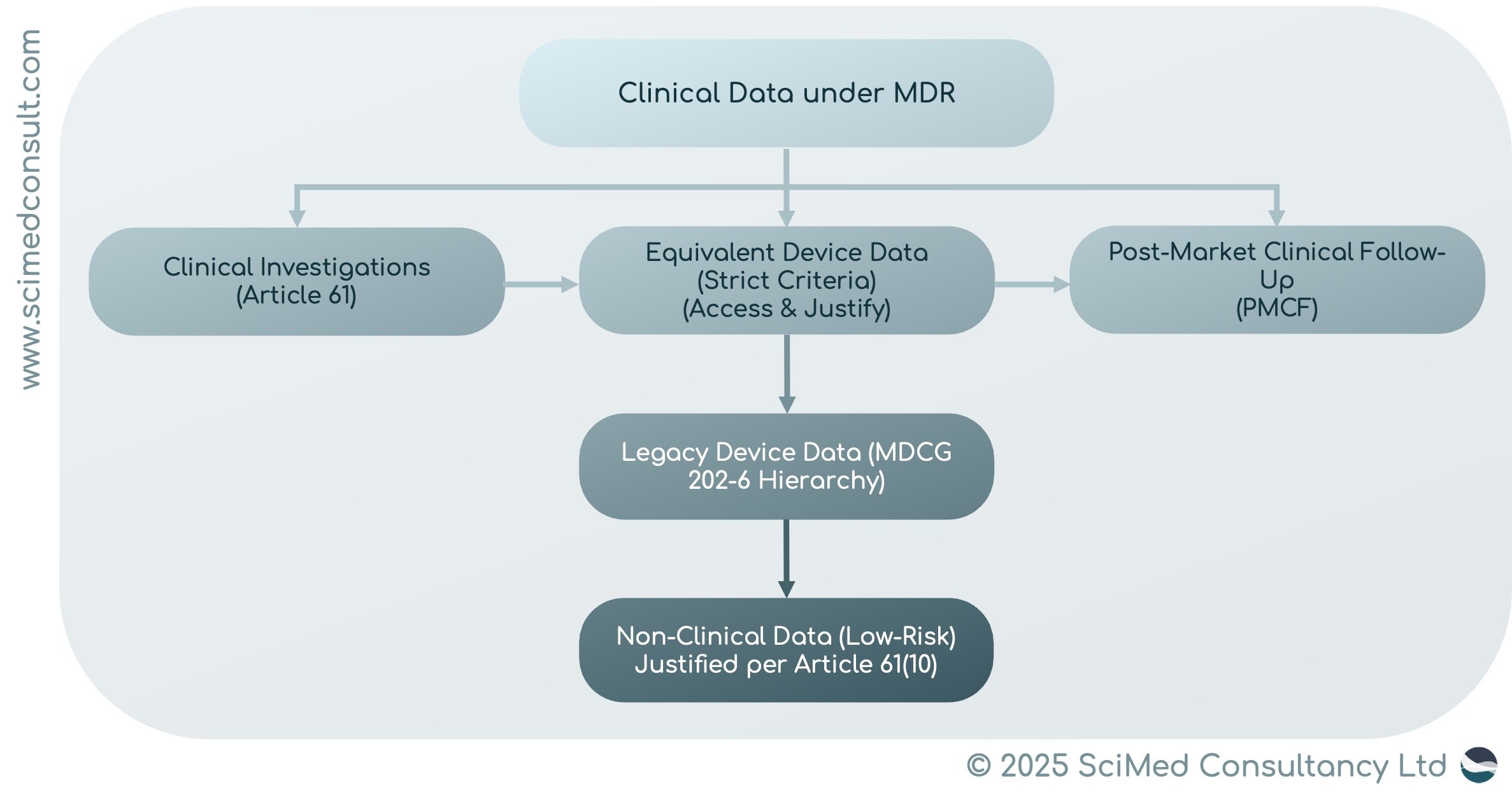
WHAT COUNTS AS CLINICAL DATA UNDER MDR
The MDR defines ‘clinical data’ in Article 2, Section 48. Acceptable sources include:
Clinical Investigations with the Device Itself
Gold standard for new devices, particularly Class III or implantables.
Clinical Investigations or Studies on Equivalent Devices
Permissible under strict equivalence criteria: technical, biological, and clinical similarity must be demonstrated, with full data access and scientific justification.
Pro Tip
Only claim equivalence if you have legally binding, full access to an equivalent's device data.
Post-Market Clinical Follow-Up (PMCF) Data
Real-world evidence from registries, surveys, and post-market studies. PMCF must address gaps identified in initial clinical evaluations.
Legacy Device-Specific Data
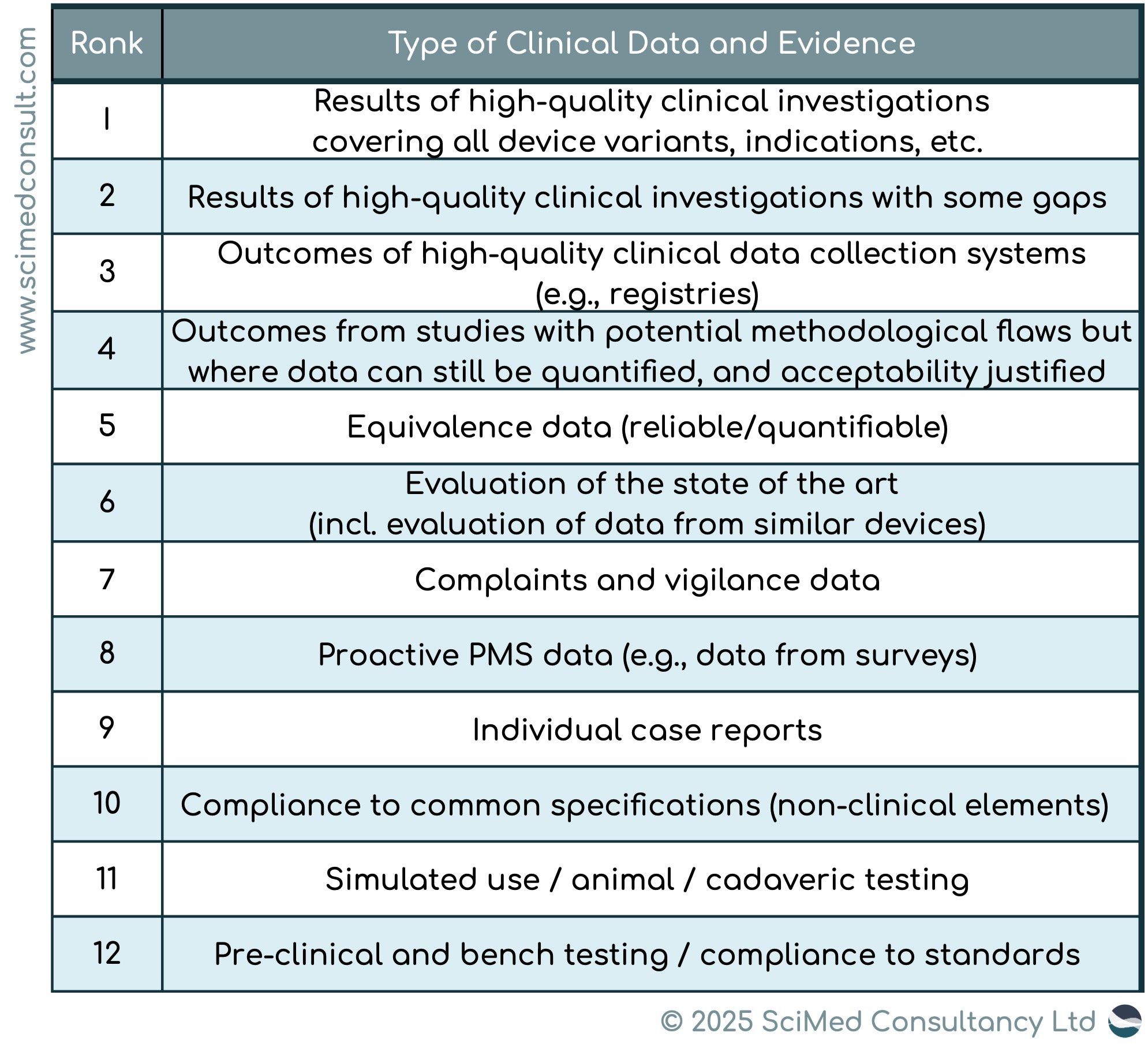
Includes scientific literature, PMS/PMCF data, registries, and cumulative evidence from multiple sources.
MDCG 2020-6 provides a hierarchy of clinical evidence for legacy devices (see below table). For example; for high risk devices, such as Class III/implantable devices, a high degree of clinical evidence is required (clinical data at a minimum of Rank 4 in the hierarchy). However, for legacy devices, the clinical evidence is lower, at Rank 6 of the hierarchy.
Non-Clinical Data
Allowed only for low-risk, non-critical devices, with strong justification.
Pro Tip
Make sure you have appropriate and sufficient quality of clinical data for your device. For example, new devices require higher levels of evidence.
WHAT DOESN’T COUNT (ANYMORE)
The MDR explicitly limits reliance on:
Equivalence without robust justification.
Outdated or indirect literature.
Manufacturer-held data without adequate disclosure.
General claims of state-of-the-art performance without supporting clinical data.
Note: Notified Bodies increasingly reject submissions relying on vague claims, poorly structured data, or insufficient justification.
WHY IT MATTERS
Regulatory Delays: Insufficient clinical evidence is a leading cause of MDR certification delays.
Increased Costs: Additional clinical investigations studies may be required to meet MDR expectations.
Market Withdrawal: Legacy devices may be removed if evidence is insufficient.
Manufacturers are expected to treat clinical evaluation as a living process, continuously updating PMCF and CERs as new data emerges.
DOES YOUR CLINICAL EVIDENCE STILL MEET MDR EXPECTATIONS IN 2026?
With evolving guidance and proposed regulatory changes, many clinical evidence strategies that were previously acceptable may no longer meet scrutiny.
Our MDR Clinical Evaluation Audit Checklist helps you assess whether your current approach will hold up under notified body review.
It helps you identify:
Reliance on outdated or insufficient clinical data
Weak or unsupported equivalence claims
Gaps between CEP, CER, and PMCF strategy
Misalignment with current MDCG expectations
Where your evidence may be challenged under updated MDR interpretation
PRACTICAL STEPS FOR MANUFACTURERS
Start early: Develop a Clinical Evaluation Plan (CEP) during device development.
Prioritize device-specific data: Conduct clinical investigations whenever possible.
Use PMCF strategically: Plan post-market studies or surveys to fill evidence gaps.
Be transparent: Fully justify equivalence data and disclose differences.
Follow guidance: Align with MDCG documents (e.g., 2020-6 for legacy devices).
Justify omissions: Clearly explain any gaps in clinical data, particularly for legacy devices.
Run your own MDR Clinical Evidence Audit
Use our structured checklist to assess whether your clinical data, CER, and PMCF approach align with MDR expectations.
Designed for regulatory teams preparing for:
Initial MDR submission
CER updates
Notified Body review
A practical tool used in real audit preparation. Not a generic template.
SUMMARY
In the MDR landscape, understanding what counts as acceptable clinical evidence is critical to avoid costly delays and ensure compliance. Manufacturers must approach clinical evaluation as an ongoing, strategic process, leveraging device-specific investigations, PMCF, and transparent equivalence claims to demonstrate conformity, safeguard patients, and maintain market access.
CLINICAL EVIDENCE EXPECTATION UNDER MDR ARE CONTINUING TO EVOLVE, PARTICULARLY AS NEW REGULATORY PROPOSALS EMERGE
MedTech Horizon provides monthly guidance on clinical evaluation, CER updates, and how manufacturers are adapting to regulatory change in practice.
If you are reviewing your clinical evidence strategy in response to recent MDR developments, you can speak with our team for a focused discussion.