How to Build a Compliant PMS System
Ensuring PMS Compliance and Audit-Ready Documentation
“A compliant PMS system is more than a collection of complaints; it combines patient safety, device performance, and risk management in an integrated and ongoing approach - proactive, not reactive.”

by Dr Nicola Edwards
Senior Analyst, SciMed Consultancy Ltd
EXECUTIVE SUMMARY
Under the EU Medical Device Regulation (EU MDR), all manufacturers must establish a proactive and systematic Post-Market Surveillance (PMS) system (Articles 83-86).
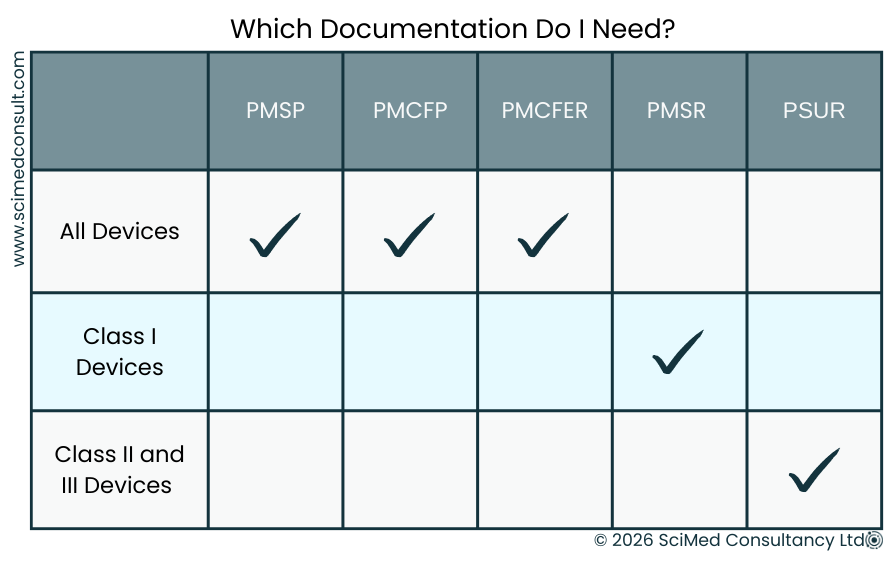
A compliant PMS framework requires a PMS Plan for all devices, supported by either a PMS Report (Class I) or a PSUR (Class IIa-III).
Post-Market Clinical Follow-Up (PMCF) activities must be planned, documented, and integrated with clinical evaluation and risk management.
PMS documentation must enable continuous reassessment of safety, performance, and benefit-risk throughout the device lifetime.
Regular updates and clearly defined methodologies are essential to demonstrate ongoing compliance.
YOUR CHECKLIST FOR COMPLIANCE
Are your surveillance methods proactive, systematic, and covering the lifetime of the device?
Do you have a PMS plan in place?
Have you checked whether you need a PMS report or a PSUR?
Do you have a PMCF plan and report in place?
Do you have regular document updates scheduled?
INTRODUCTION
The EU Medical Device Regulation (EU MDR) defines post-market surveillance as ‘all activities carried out by manufacturers in cooperation with other economic operators to institute and keep up to date a systematic procedure to proactively collect and review experience gained from devices they place on the market, make available on the market or put into service for the purpose of identifying any need to immediately apply any necessary corrective or preventive actions’. Article 83 further emphasises the importance of a comprehensive PMS system in monitoring the safety and performance of a device throughout its lifetime, with results feeding into key documentation such as clinical evaluation and risk management.
Crucially, building a compliant PMS system requires more than simply collecting complaints or reacting to incidents. The system must be proactive, systematic, and continuously maintained. It should enable manufacturers to identify trends as they emerge, reassess benefit-risk where necessary, and implement corrective or preventive actions in a timely manner.
This article outlines the key documentation required to establish and maintain a compliant PMS system under EU MDR, including the PMS Plan, PMS Report or PSUR, and PMCF documentation, and explains how these elements work together to support ongoing regulatory compliance throughout the device lifecycle.
INDUSTRY CONTEXT & BACKGROUND
Post-market surveillance requirements have evolved significantly with the transition from the Medical Devices Directive (MDD) to the EU Medical Device Regulation (MDR). Under the MDD, PMS activities were largely reactive, focused on complaint handling and adverse event reporting. MDR, by contrast, establishes a proactive, systematic, and lifecycle-based approach, requiring manufacturers to continuously monitor device safety, performance, and benefit-risk across the entire intended lifetime of their devices.
This shift reflects the European Commission’s increased emphasis on patient safety, real-world evidence, and regulatory oversight. Notified Bodies are now scrutinising PMS and PMCF documentation more rigorously, paying particular attention to:
The robustness and traceability of data collection and analysis methods
Thresholds and indicators that trigger corrective or preventive actions
Integration between PMS outputs, clinical evaluation reports (CERs), and risk management files
STAYING ALIGNED WITH PMS REQUIREMENTS UNDER MDR
Post-market surveillance expectations continue to evolve, particularly around PMCF integration, real-world evidence, and audit readiness.
MedTech Horizon provides monthly guidance on post-market surveillance, clinical evaluation updates, and how manufacturers are maintaining compliance in practice.
For manufacturers, this means that PMS is no longer an isolated regulatory activity. It must be integrated with PMCF, vigilance, and risk management processes, and tailored to device class, intended use, patient population, and market characteristics. A well-designed PMS system not only ensures compliance but also provides actionable insights into device performance, supporting informed decision-making, ongoing safety monitoring, and sustained regulatory confidence.
The PMS Plan
The key document that establishes the PMS system is the PMS Plan, produced in accordance with MDR Article 84, with MDR Annex III providing more detail on content. All manufacturers must produce a PMS Plan, and Class I devices must also have a PMS Report (Article 85).
The PMS Plan defines the methodology for data collection, whereas the PMS Report reflects on the outcomes of these activities.
The following information should be outlined in the PMS Plan and discussed in the PMS Report:
Serious incidents, field safety corrective actions, and information from PSUR documentation
Non-serious incidents and undesirable side effects
Trend reporting
Relevant specialist or technical literature, databases, or registers
Information provided by users, distributors, or importers (e.g., complaints, feedback)
Publicly available information about similar devices
The PMS Plan must therefore:
Describe effective and appropriate methods for PMS data collection and assessment
Include thresholds and indicators for reassessing benefit-risk and risk management
Provide methods and protocols to follow in the event of a statistically significant increase in frequency or severity of incidents, the need for corrective action, or necessary communication with competent authorities
Link to a PMCF Plan for ongoing clinical follow-up activities
Pro Tip
Ensure traceability between PMS findings, risk management actions, and clinical evaluation updates to demonstrate proactive, systematic surveillance during audits.
The PSUR
A Periodic Safety Update Report (PSUR) must be generated for all Class IIa, IIb, and III devices, in accordance with Article 86 of the MDR. The PSUR summarizes the PMS data gathered but provides greater detail than a PMS Report. Guidance on content is outlined in MDCG 2022-21.
A compliant PSUR should include all elements of the PMS Report, plus:
Conclusions of the benefit-risk determination
Main findings of PMCF activities
Device sales volume, estimated user population, and device usage frequency
PSURs should cover the intended lifetime of the device, and be updated at least every two years for Class IIa, and annually for Class IIb or III.
Pro Tip
Document trends and emerging risks clearly; this demonstrates a proactive approach to regulators.
The PMCF Activities
Post-market clinical follow-up activities are conducted proactively using the CE-marked device in a relevant human population. PMCF is outlined in MDR Annex XIV Part B, with templates in MDCG 2020-7 and 2020-8.
PMCF activities are guided by a PMCF Plan, with results analysed in a PMCF Evaluation Report. The three key aims of these documents are:
Confirm the safety and performance of the device throughout its lifetime, ensuring the benefit-risk ratio remains acceptable
Identify and monitor side effects and emergent risks
Verify that the intended purpose is correct by identifying possible misuse or off-label use
The PMCF Plan should include:
Methods and applications of each PMCF activity (e.g., user feedback, literature searches)
Rationale for appropriateness of methods and procedures
Specific objectives for PMCF activities
Detailed and justified time schedule for each activity
Evaluation of data relating to similar or equivalent devices
References to relevant parts of the clinical evaluation report and risk management documentation
References to common specifications, harmonised standards, and guidance documents
Pro Tip
Ensure PMCF objectives and methods are clearly linked to CER updates and risk management to satisfy Notified Body expectations.
CONCLUSION
A compliant PMS system is proactive, systematic, and regularly updated. Its documentation consists of a PMS Plan, PMCF activities, and a PMS Report or PSUR, which together assess safety, performance, and benefit-risk outcomes over the intended lifetime of a device.
The conclusions made in these documents are based on robust clinical evidence collected from multiple sources, ensuring that surveillance activities are not only compliant but actionable for risk management and product lifecycle decisions.
If you are reviewing your PMS system or preparing for audit, you can speak with our team for a focused discussion.