EU MDR Post-Market Surveillance: Building a Continuously Audit-Ready Evidence System
“Most MDR audit findings I see now sit at the joins between documents. The PMS report, the CER, the risk file and the PSUR are usually well written; the connective tissue between them is where surveillance systems give way under scrutiny.”

Alastair Selby
Managing Director, SciMed Consultancy Ltd
Executive Summary
MDR has transformed post-market surveillance from a periodic compliance activity into a continuously operating evidence system.
Notified Bodies increasingly assess the consistency and traceability between PMS, PMCF, CER, Risk Management and PSUR documentation.
Many audit findings arise not from missing documents, but from disconnected evidence flows and inconsistent conclusions across files.
State-of-the-art monitoring is now expected to operate continuously, with defined surveillance methodologies and documented escalation pathways.
Sustainable MDR compliance depends on governance, integration workflows and operational systems — not isolated document refresh cycles.
Introduction
The audit findings stacking up under MDR post-market surveillance share a recurring character: they involve documents that exist but disagree with each other. A PMS report that lists complaints the CER never references. A risk file that hasn't moved since the last technical documentation submission. A PSUR that quotes a benefit-risk conclusion the underlying clinical data no longer supports. The deliverables are all in the QMS, individually competent, collectively incoherent.
This is the operational reality manufacturers are living with several years into MDR enforcement, and it's the issue Notified Bodies have started to scrutinise hardest now the industry has transitioned from implementation to maintenance. Reviewers now assess surveillance as a system: how the data flows, how findings travel between files, how conclusions stay coherent over time, and whether the evidence behind any single claim can still be reconstructed when a reviewer asks.
The manufacturers coping well have stopped treating PMS, PMCF, CER, Risk, PSUR and SSCP as separate workstreams and started running them as a single, continuously updated evidence ecosystem. The shift from documentation programme to evidence lifecycle is what this article is about.
We'll cover why PMS has become the most operationally exposed area under MDR, why apparently compliant systems still fail at audit, what reviewers actually look for, and what a sustainable, audit-ready operating model looks like in practice.
Why Post-Market Surveillance Has Become a Major Operational Risk Area Under MDR
Under the AIMDD and MDD regimes, post-market surveillance was, for most manufacturers, a periodic exercise. A vigilance system, a complaint log, an annual review, a refresh of the CER every few years. The regulatory weight sat firmly on the pre-market side.
MDR has reversed that. Articles 83 to 86, together with Annex III, have made PMS the active spine of conformity. The plan, the report, the PSUR and (where applicable) the SSCP have become outputs of a system that has to ingest data continuously and update conclusions when that data shifts. PMCF (Annex XIV Part B) extends the same logic to clinical evidence, requiring proactive, structured generation of post-market clinical data unless properly justified otherwise.
The operational consequence is that surveillance now demands more sustained attention than any other area of compliance. CER content updates flow from PMS findings. Risk files have to absorb new hazards, new severity data, and revised state-of-the-art positions. PSURs have to reconcile complaints, vigilance signals, PMCF results, literature, and benefit-risk conclusions in a way that's coherent with everything else in the technical documentation. Every input changes the picture. Nothing stands still.
For Regulatory Affairs and Quality leaders, this has translated into a quiet but persistent operational risk. The system never finishes. Every quarter brings new complaints, new literature, new field actions in the wider device class, new regulatory expectations on state-of-the-art monitoring. Teams sized for periodic compliance are being asked to maintain a continuous one. The gap between the workload PMS now generates and the resources available to absorb it is, in our experience, the single most common source of audit exposure under MDR.
Why Many PMS Systems Fail Under Notified Body Scrutiny
Audit findings on PMS rarely come from low-quality documentation. The documents themselves are usually competently written. What fails is the system underneath them.
The pattern we see most often is one of plausible isolation. The PMS plan is well drafted, the PMS report is up to date, the PSUR is on schedule. Each artefact looks credible on its own. The problem appears the moment a reviewer pulls a thread between them. A complaint trend in the PMS report doesn't reach the risk file. A literature finding in the CER hasn't been weighed in the PSUR's benefit-risk conclusion. A PMCF interim result hasn't fed back into the PMS plan as an updated indicator. Each document tells the truth about its own slice of the picture, and together they produce a narrative that doesn't quite hold.
Notified Bodies have become noticeably better at finding these inconsistencies. Reviewers now routinely cross-walk between PMS, PMCF, CER, Risk and PSUR, looking specifically for points where a finding in one document should have triggered a change in another and didn't. When that linkage breaks, even a beautifully written PSUR becomes a finding.
The other recurring failure mode is temporal. Documents are technically current but materially stale: a risk file dated this year that still cites state-of-the-art positions from three years ago, a CER refresh that hasn't pulled in the most recent six quarters of complaint data, a PSUR conclusion repeated from the prior cycle without genuine reanalysis. Reviewers are increasingly explicit that being in date is one thing and being current is another. They want evidence that the conclusions reflect the data as it stood at the moment the document was signed.
The cause sits in the operating model. PMS has been treated as a set of recurring deliverables, and recurring deliverables, no matter how well written, drift apart over time when the underlying system that should connect them is left to ad hoc effort.
In practice, most post-market system weaknesses emerge not from a complete absence of documentation, but from gradual fragmentation across interconnected evidence activities.
| Failure Point | What It Looks Like in Practice | Operational Consequence |
|---|---|---|
| Fragmented Evidence | PMS, PMCF, CER, PSUR and risk documentation are maintained independently with limited cross-referencing or traceability. | Inconsistent narratives, reviewer confusion, increased audit findings and remediation effort. |
| Manual Surveillance Burden | Literature reviews, vigilance monitoring and signal detection activities rely heavily on manual tracking and disconnected spreadsheets. | Resource strain, delayed updates, inconsistent surveillance cadence and reduced oversight confidence. |
| Weak PMS → CER Integration | PMS findings are not consistently reflected in CER conclusions, benefit-risk updates or clinical evidence discussions. | Defensibility issues during Notified Body review and difficulty justifying ongoing safety and performance conclusions. |
| Inconsistent Benefit-Risk Narratives | Different post-market deliverables present conflicting conclusions, rationales or risk interpretations over time. | Reduced reviewer confidence and increased likelihood of nonconformities or follow-up questions. |
| Lack of Lifecycle Governance | No clear ownership, update cadence or integrated oversight across post-market activities. | Reactive remediation cycles, operational inefficiency and recurring audit preparation stress. |
Key Message
These issues rarely emerge from a complete absence of documentation.
More commonly, they arise when evidence evolves in parallel rather than through a continuously maintained and traceable lifecycle system.
These issues rarely appear in isolation. Once evidence continuity begins to weaken, organisations often experience increasing remediation workload, reviewer queries, and operational strain during subsequent update cycles.
Audit Insight
If a reviewer follows a signal from complaint data into your CER and Risk File, the narrative and conclusions within each should align without manual explanation.
Running the PMS Plan as Live Infrastructure
The mental model that quietly underpins most struggling PMS programmes is the document model. PMS plan, PMS report, PSUR, SSCP, each with an owner, a template, and a due date. Compliance is conceived as the act of producing each artefact on time.
Under MDR, that model breaks down. Annex III makes the PMS plan an instrument that must be capable of absorbing change. Indicators have to be defined in a way that lets the manufacturer detect emerging trends. Threshold criteria have to actually trigger something. The plan itself has to evolve as the data evolves. The PMS report and PSUR are expected to reflect a current view of the device's safety and performance at the moment of signing, supported by the underlying data as it then stood.
In practice this means the PMS plan should be running as live infrastructure. The indicators it defines have to map to data the organisation can actually capture, at the cadence the plan claims. The trend analysis methodology has to produce outputs the team can review and act on. The link between PMS data and the risk file has to be operational, with documented decision points. The interface with PMCF activities has to be specified clearly enough that a reviewer can follow what surveillance data triggered a PMCF update, and how.
The surest route to incoherence is to write the plan once at certification and refresh it only when audited. Manufacturers that handle this well run the plan as the operating manual for their surveillance system, with version control, periodic governance review, and explicit linkage to the underlying QMS procedures. Everything downstream becomes easier to write because the system that feeds it is already coherent.
Post-market activities can no longer operate as isolated updates performed independently across teams and deliverables. Mature systems rely on continuous evidence flow between PMS, PMCF, CER maintenance, risk management, PSUR activities, and ongoing surveillance processes.
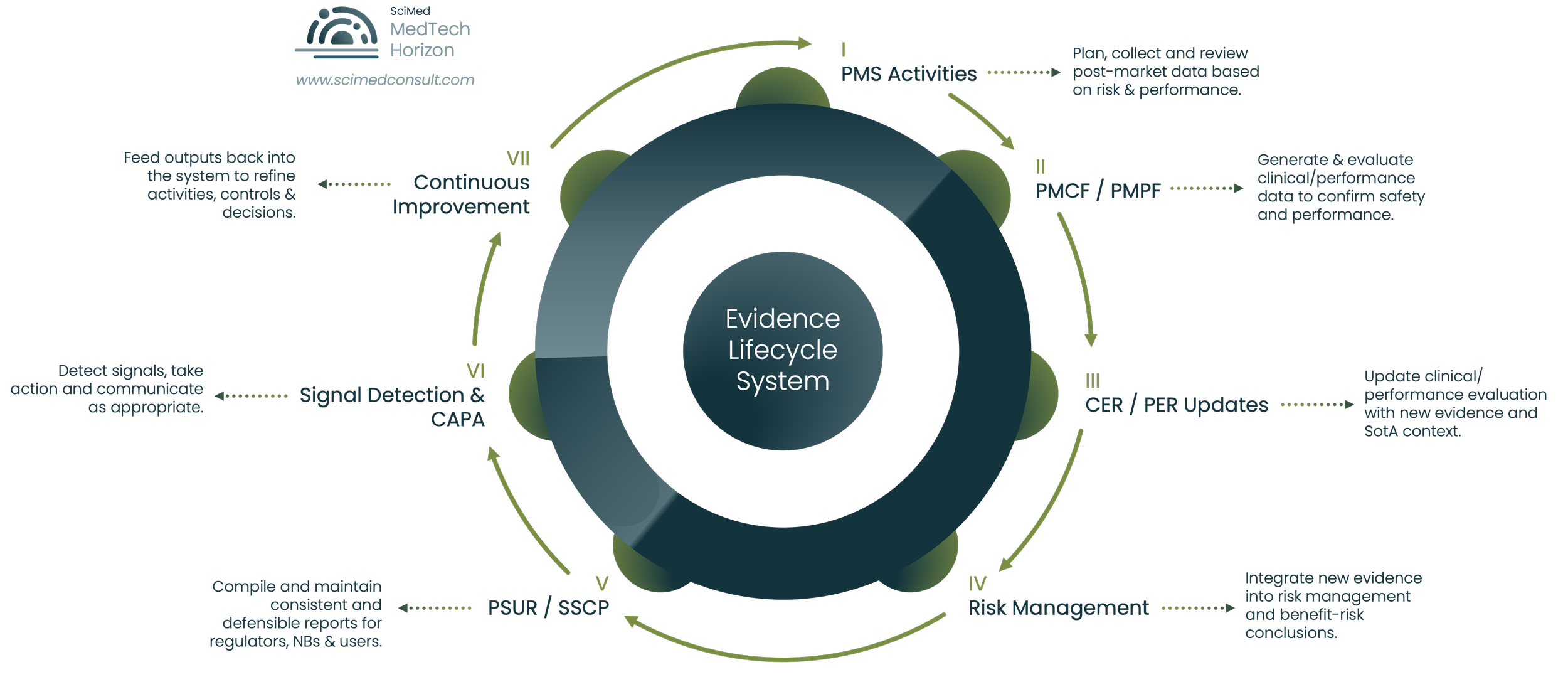
Figure 1.
The MDR Evidence Lifecycle System - illustrating how PMS, PMCF, CER/PER maintenance, risk management, PSUR/SSCP activities, and continuous improvement processes function as an interconnected post-market evidence ecosystem rather than isolated regulatory deliverables.
Pro-Tip
Treat your PMS Plan as a live operational framework, not a certification deliverable that only changes during audits.
The Hidden Problem: Evidence Fragmentation
Evidence fragmentation is the issue most manufacturers underestimate, partly because each individual fragment looks fine. Complaints sit in a complaints database. Vigilance reports sit with the vigilance lead. PMCF data sits with clinical. Literature surveillance sits with the CER author or an external partner. Field action data sits with QA. Risk files sit with the engineering or safety function. Each repository is competently maintained.
The fragmentation lives in the connective tissue between sources, where data has to move between functions, get reconciled, and update related artefacts. There's no single point at which all relevant evidence is brought together, weighed, and reconciled into a current view of the device. The PSUR is supposed to be that point. In fragmented systems the PSUR is written by extracting from each silo at a single moment in time. The ongoing integration of those sources, which is what the regulation assumes, doesn't happen as a matter of process.
The consequences are predictable. Findings move slowly between functions, sometimes failing to move at all. A complaint pattern noticed by the customer service team takes months to appear in the risk file. A literature finding flagged by the clinical team doesn't reach PMS until the next periodic review. A PMCF observation that should have changed an indicator in the PMS plan instead sits in a clinical study report. Each handoff is a place where evidence loses traceability.
The downstream effect is that, when a reviewer asks the simple question,
"Show me how this conclusion was reached,"
the answer has to be reconstructed across multiple systems and multiple owners. The reconstruction is usually possible, just slow, error-prone, and visible. Reviewers notice when teams can't produce evidence trails on demand.
The mature alternative makes the evidence thread visible by design. A defined integration model is what does the work. Named owners on each side of every interface, documented handoffs, decision points captured at the time they're made, and a governance layer that ensures findings travel between domains within agreed timeframes. The platform choice matters less than the integration model itself. The integration work that fragmented systems leave to ad hoc effort has to be built into the operating model.
The MDR Evidence Thread: PMS, PMCF, CER, Risk Management & PSUR Integration
The phrase that keeps coming up in our work is the evidence thread. It captures what an audit-ready system looks like operationally: a single, traceable line of evidence that runs through every regulatory artefact, so that a finding entering the system at any point can be followed all the way through to its impact on the benefit-risk conclusion.
PMS → PMCF → CER → Risk → PSUR → SSCP
The thread runs roughly in this sequence, though in practice the loops go both ways:
PMS as the Entry Point for Real-World Evidence
PMS captures and analyses real-world data: complaints, vigilance signals, registry data, user feedback, service data, and broader market intelligence on the device class. It's the entry point for almost everything the rest of the system has to react to.
How PMCF Extends the Clinical Evidence Base
PMCF generates structured, proactive clinical evidence to confirm that performance and safety hold up across the indicated population over the device's lifetime. PMCF activities should be planned in response to PMS findings as well as pre-market gaps. The PMS plan and the PMCF plan should be visibly related documents, drafted in conversation with each other.
Integrating PMS and PMCF into the Clinical Evaluation
CER integrates PMS and PMCF outputs with literature and other clinical data into the current clinical evidence base. Each periodic CER update should make explicit reference to what's changed in PMS and PMCF since the last version, and what that change means for the clinical evaluation conclusion.
Maintaining Current Risk Management Under ISO 14971
Risk management ingests the same evidence to update hazard analyses, severity and probability estimates, residual risk, and benefit-risk position. ISO 14971 and the regulation are explicit that risk has to remain current. In practice, risk only stays current if PMS and PMCF feed it on a defined cadence rather than at periodic refresh points.
PSUR as the Evidence Integration Layer
PSUR (or the PMS report for lower-risk devices) is the integration point. It pulls together everything: complaints, vigilance, PMCF, literature, risk updates, state-of-the-art changes, and produces a current benefit-risk statement that is internally consistent with the CER and risk file.
Maintaining SSCP Consistency
SSCP, where applicable, is the public-facing summary, and it has to remain consistent with all of the above.
With this thread intact, audits move differently. The reviewer asks how a specific complaint pattern affected benefit-risk, and the team can show the path from the complaint database, through PMS analysis, into the risk file, through to the PSUR conclusion and the corresponding CER update. With it broken, that path has to be reconstructed under pressure, and reconstructed paths leak.
Operational Insight
The longer evidence takes to move between PMS, PMCF, Risk and CER activities, the greater the likelihood of audit-visible inconsistencies.
What Notified Bodies Actually Look For
The regulation specifies what's required. What reviewers actually focus on in practice, is a smaller and more diagnostic set of patterns. Three dominate.
The first is coherence under cross-examination.
Reviewers will pick a specific signal, an adverse event, a complaint cluster, a piece of recent literature, and follow it across documents. They want to see that the same signal is acknowledged consistently across PMS, risk, CER and PSUR, and that its weight in the benefit-risk conclusion is justified the same way in each place. Inconsistency in framing or quantification, even where each individual mention is technically defensible, draws attention.
The second is responsiveness over time.
Reviewers look at the gap between when information became available and when it influenced the regulatory documentation. A literature finding from eighteen months ago that's only now being addressed in an upcoming PSUR raises the question of how the system catches things in between. The expectation is that the cadence at which evidence flows into the documentation is shorter than the formal review cycle of any individual document.
The third is the credibility of the methodology.
PMS plans that define indicators in vague qualitative terms, trend analyses that don't actually compute trends, state-of-the-art monitoring that consists of one search a year. All of these are treated more critically than they were a few years ago. Reviewers want methodology capable of detecting a problem if a problem were present, with thresholds, time horizons and comparator baselines specified clearly enough that the analysis could fail informatively.
Underneath all three is a question of trust. The reviewer is, in effect, asking whether the surveillance system is capable of producing reliable answers. Where the cross-walks line up, response times are short, and the methods are sound, the rest of the audit usually moves quickly. Where they don't, the audit shifts into a deeper investigation of how the system works, and that's where findings accumulate.
Run Your Own MDR PMS Audit-Readiness Review
Most PMS systems do not fail because documents are missing.
They fail because evidence no longer moves coherently between PMS, PMCF, CER, Risk Management and PSUR activities.
Our MDR PMS Audit Readiness & Evidence Traceability Checklist helps regulatory and quality teams assess:
PMS → CER evidence flow
PMCF integration gaps
Risk-management alignment
PSUR consistency
State-of-the-art monitoring maturity
Audit-visible fragmentation risks
Lifecycle governance weaknesses
Get the checklist instantly as a professionally formatted PDF.
Used by MedTech manufacturers preparing for MDR surveillance audits, CER updates, and lifecycle remediation activities.
Common PMS System Failure Points
Across the manufacturers we work with, the failure points cluster into a small number of recurring patterns. Recognising them early is usually cheaper than addressing them at audit.
Indicator Sprawl
Indicator sprawl is one. PMS plans accumulate indicators over time without a corresponding methodology for reviewing them. The plan looks thorough on paper but the team can't actually report against half the indicators in the way the plan describes. At audit, the gap between plan and execution becomes visible.
Threshold Criteria That Never Trigger Action
Threshold criteria that don't trigger anything is another. The plan defines what counts as a signal, but the procedure for what happens when a signal is detected is either missing or so generic it adds no value. Reviewers ask what happened the last time a threshold was breached. If the answer is "we noted it", that's a finding.
Tickbox State-of-the-Art Monitoring
State-of-the-art monitoring done as a tickbox is a third. A literature search at CER refresh time, with no continuous mechanism to detect changes in standards, medical alternatives, or clinical practice in between. MDR's expectations on state-of-the-art are not met by an annual exercise.
PMCF Activities That Fail to Close the Loop
PMCF integration that doesn't close the loop is a fourth. PMCF activities run, results emerge, but there's no documented mechanism by which those results update the PMS plan, the risk file, or the CER. The PMCF report becomes a deliverable in its own right, divorced from the rest of the system.
Risk Files Frozen at Certification
Risk files frozen at certification is a fifth, and probably the most common. The risk management file was thoroughly updated for the technical documentation submission and hasn't materially moved since. PMS data has been collected, but the risk file hasn't absorbed it. By the time the next CER cycle comes around, the gap is large enough to be visible.
Governance Gaps Between Functions
Governance gaps complete the picture. There's no defined forum at which PMS, clinical, risk, and regulatory functions meet to reconcile findings and agree downstream actions. The integration the regulation assumes will happen is left to ad hoc coordination, which works when teams are quiet and breaks under pressure.
The cause, again, is structural. The integration that the regulation assumes will happen has no defined place to happen, so it doesn't, until pressure forces it.
Pro-Tip
Trend indicators only add value if the organisation can realistically collect, review and act on the data they generate.
State-of-the-Art Monitoring Under MDR: The Continuous Evidence Burden
State-of-the-art surveillance is the area where the gap between regulatory expectation and operational capacity is widest. MDR expects manufacturers to maintain a current view of clinical practice, applicable standards, medical alternatives, and emerging safety information across the device class. Notified Body guidance has reinforced this, and reviewers are increasingly probing how state-of-the-art is monitored continuously rather than periodically.
One of the most underestimated aspects of MDR post-market compliance is the cumulative cadence burden created by overlapping surveillance, reporting, evaluation and update obligations.
| Deliverable | Applies To | Regulatory Reference | Typical Frequency | Strategic Purpose |
|---|---|---|---|---|
| PMS Plan | All MDR devices | Annex III | Established initially and maintained continuously | Defines the structure, methods and responsibilities for PMS activities. |
| PMS Report | Class I devices | Article 85 | Updated as necessary | Summarises PMS findings and conclusions for lower-risk devices. |
| PSUR | Class IIa devices | Article 86 | At least every 2 years | Maintains ongoing assessment of benefit-risk profile and PMS outcomes. |
| PSUR | Class IIb and III devices | Article 86 | At least annually | Demonstrates continuous evaluation of safety, performance and post-market evidence. |
| SSCP | Implantable and Class III devices | Article 32 | Reviewed and updated regularly | Provides publicly accessible summary of safety and clinical performance. |
| PMCF Evaluation Report | Devices requiring PMCF | Annex XIV Part B | Typically annual or aligned to CER/PSUR cycle | Confirms continued safety, performance and clinical benefit. |
| CER / PER Update | MDR and IVDR devices | Annex XIV / IVDR Annex XIII | Based on device class, risk profile and PMS findings | Maintains defensible clinical or performance evaluation conclusions. |
| State-of-the-Art Review | All evidence-based evaluations | MDCG and MEDDEV expectations | Continuous or periodic surveillance | Ensures conclusions remain aligned with current clinical and technological context. |
| Risk Management File Update | All MDR devices | ISO 14971 / Annex I | Continuous lifecycle activity | Integrates new PMS findings into benefit-risk management. |
When these activities are managed independently rather than through a coordinated lifecycle system, operational strain and evidence inconsistency increase significantly over time.
The operational implication is that some part of the organisation has to be reading regularly. New literature, new versions of harmonised standards, new common specifications, new MDCG documents, Team-NB position papers, MHRA and FDA communications, registry outputs, professional society guidance, recall data on comparable devices. None of this is individually overwhelming. Cumulatively, across a portfolio, it's substantial.
One of the most underestimated aspects of MDR post-market compliance is the cumulative workload created by overlapping surveillance, review, reporting, and update obligations operating on different cadences simultaneously.
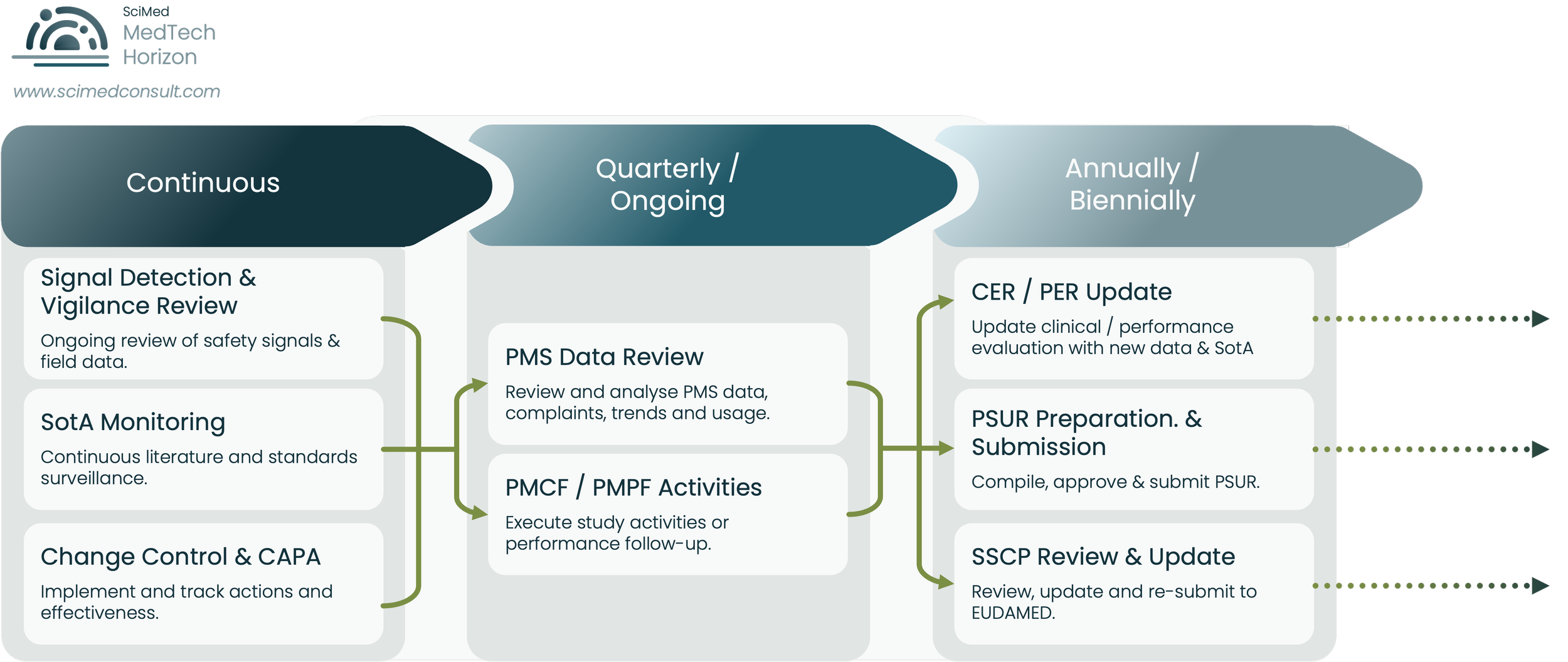
Figure 2.
Illustrative example of the overlapping cadence burden created by continuous surveillance activities, periodic PMS review processes, and recurring MDR post-market reporting obligations across the evidence lifecycle.
Manufacturers who manage this without burning their teams out tend to do three things consistently.
They define the watch list explicitly. Standards in scope, journals in scope, regulatory and competent authority feeds in scope, comparator devices in scope, and clinical practice domains in scope. The watch list is documented, owned, and revisited periodically.
They run surveillance on a defined cadence. Monthly literature scans for high-risk devices, quarterly broader reviews, annual full-portfolio updates feeding the CER refresh. The cadence is short enough that nothing material can sit undetected for a full review cycle.
They link findings to actions through a single intake point. Whatever the watcher finds, it lands in a structured log with categorisation, impact assessment, and routing to the relevant owner: risk, clinical, PMS, regulatory. The log itself becomes evidence at audit that the system is operational.
State-of-the-art treated as a CER appendix exercise is now the area reviewers scrutinise hardest, and where findings accumulate fastest.
Compliance Insight
State-of-the-art monitoring should have defined ownership, cadence and escalation criteria, not rely on annual CER refresh cycles.
Building a Sustainable, Audit-Ready PMS Operating System
Sustainability is the right word here. The failure mode that catches manufacturers out under MDR is the inability to keep producing coherent documents in alignment with each other over years, with the system increasingly held together by individual effort. The operating model has to be designed for that.
A workable architecture has four layers:
The first is the Data Layer.
Defined sources, defined fields, defined collection cadences. Complaints, vigilance, service data, PMCF outputs, literature, registry data, market intelligence on comparators. Each source has an owner, a quality standard, and a defined route into the surveillance system. Without this layer, every downstream document is built on shifting foundations.
The second is the Analysis Layer.
PMS plan indicators, trend analysis methodology, risk re-evaluation triggers, benefit-risk reanalysis criteria. This is where data becomes information and where most struggling systems are weakest. Methodology that could plausibly detect a real-world problem is the standard to design to.
The third is the Integration Layer.
The defined points at which findings move between domains: PMS to risk, PMS to CER, PMCF to PMS, literature to risk, state-of-the-art to all. Each interface has a named owner on each side, a defined trigger, a defined handoff format, and a defined timeline. Governance, in the form of a periodic cross-functional review, sits over this layer to catch things that fall between interfaces.
The fourth is the Documentation Layer.
PMS plan, PMS report, PSUR, CER updates, risk file revisions, SSCP. The documentation layer is downstream of the first three. Working harder on documents to compensate for weakness lower down is expensive and doesn't hold under audit.
Building this is mostly a matter of definition and discipline rather than tooling, though good tooling helps. Define the operating model explicitly, write it down in QMS procedures, allocate ownership, and run governance against it. Once the operating model is in place, individual deliverables become much faster to produce and much easier to defend, because they're outputs of a coherent system.
The distinction between reactive and audit-ready post-market systems is rarely about effort alone. More often, it reflects whether evidence activities are governed as isolated updates or as part of a continuously maintained lifecycle framework.
| Reactive PMS System | Audit-Ready Evidence System |
|---|---|
| Annual panic-driven updates | Continuous maintenance with a defined operational cadence |
| Siloed PMS, PMCF and CER activities | Integrated evidence lifecycle with traceable data flow |
| Manual literature surveillance and tracking | Structured, repeatable surveillance processes |
| Inconsistent benefit-risk narratives across deliverables | Consistent and scientifically defensible conclusions across outputs |
| Last-minute remediation before audits | Ongoing readiness supported by lifecycle governance |
| Update responsibility spread informally across teams | Clearly defined ownership and accountability structures |
| Limited visibility of evidence dependencies | Transparent evidence traceability across lifecycle activities |
| Reviewer queries addressed reactively | Evidence prepared with reviewer defensibility in mind |
| Operational strain increases with each update cycle | Sustainable maintenance model with controlled workload |
| Compliance viewed as periodic documentation work | Compliance managed as a continuously evolving evidence ecosystem |
Key Message
The objective is not simply to produce more documentation.
The objective is to maintain a post-market evidence system that remains coherent, traceable and defensible over time.
For many manufacturers, the challenge is not producing individual outputs. It is sustaining operational consistency across them as surveillance obligations, update cycles, and reviewer expectations evolve over time.
When MDR PMS Workloads Overwhelm Internal Teams
Most regulatory and quality teams under MDR are doing a competent job under workloads that weren't designed for continuous compliance. The signs of overload are predictable: deadlines met but with shrinking margin, increasing reliance on individual heroics, drift in the depth of analysis between cycles, postponed risk file updates, PMCF activities that slip, state-of-the-art monitoring that quietly contracts to a literature search. None of these failures are dramatic in any single instance. Cumulatively, they erode the coherence of the evidence system.
The honest test is whether the team could currently respond, in days, to a Notified Body request for the full evidence trail behind a specific benefit-risk conclusion. If the answer requires several weeks of reconstruction, the team is functionally overloaded, regardless of whether documents are nominally on schedule.
At that point, the realistic choice is rarely a binary one between fully internal and fully outsourced. The more sustainable model tends to keep internal ownership of governance, decisions, and the regulatory voice, while bringing in specialist support for the parts of the system that benefit most from external scientific and methodological depth: state-of-the-art monitoring, literature surveillance, risk-clinical integration, and PSUR/CER drafting that has to reconcile multiple data streams. Done well, this reduces the manual workload without surrendering control of the evidence narrative.
Outsourcing individual documents in isolation tends to recreate the fragmentation problem at higher cost. The integration has to be designed for, whether the work is done internally, externally, or in combination.
Need an Independent View of Your PMS System?
Many manufacturers only discover evidence fragmentation and lifecycle governance gaps when a Notified Body reviewer follows the thread between PMS, PMCF, CER and Risk Management activities.
SciMed supports manufacturers with:
PMS system remediation
CER and PSUR integration
PMCF strategy and governance
State-of-the-art surveillance frameworks
Evidence traceability reviews
Continuous audit-readiness support
Advisory-led discussions for manufacturers managing MDR post-market evidence systems.
Strategic Insight
Continuous audit readiness is ultimately an operational discipline, not a documentation exercise.
What a Mature Evidence Lifecycle System Looks Like
It helps to be concrete about what good looks like, because the gap between an MDR-compliant operating model and a typical one isn't always obvious from the outside.
A mature evidence lifecycle system has a few characteristic features:
The PMS plan reads as an operating manual.
Indicators are mapped to data sources the team genuinely captures. Thresholds are tied to defined response procedures. The plan is versioned in line with portfolio and field changes, not at fixed intervals.
Findings travel between domains within defined service levels.
A literature finding flagged in week one is logged, assessed, routed and acted on within weeks. The audit trail of that movement is itself a deliverable, available on request.
Risk files are visibly current.
Last-update dates correspond to recent surveillance activity. Hazard tables show entries that have been added, modified or retired since the last technical documentation review. Benefit-risk reasoning references current state-of-the-art and current PMS data.
PSURs and CER updates are written from a current, integrated evidence base.
The work of writing them is largely synthesis and interpretation, because the underlying inputs are already coherent.
Governance is documented and visible.
Cross-functional reviews happen on a defined cadence, with minutes, decisions and action tracking. The governance log is itself evidence that the system is operational.
State-of-the-art is monitored
Continuously, with a documented watch list, defined cadence, and a single intake point for findings. It produces a running output, with a continuous record of what was found, when, and how it was handled.
Above all, the system feels like a system. A new joiner can be shown the operating model and understand how data flows from the field, through analysis, into decisions, into documentation, in a way they can describe back. When that's true, audits move quickly, fire drills disappear, and the team's time shifts from defending isolated documents to maintaining a coherent evidence base.
Conclusion
Several years into MDR, the manufacturers pulling ahead are running surveillance as an evidence system: integrated, traceable, continuously updated, and designed to keep its conclusions coherent over years.
That shift is not free. It requires investment in the operating model, in governance, in cross-functional integration, and often in specialist support for the deepest scientific and methodological work. Continuing with periodic compliance held together by individual effort is the alternative, and it's becoming progressively more exposed as Notified Body scrutiny sharpens and state-of-the-art expectations rise. The cost of fragmentation, in audit findings, in remediation cycles, and in management distraction, eventually exceeds the cost of building the system properly.
For teams reviewing their own PMS posture, the questions worth asking are simple. Could you, today, produce the full evidence trail behind a specific benefit-risk conclusion in days rather than weeks? Are findings in your PMS data visibly reflected in your risk file and CER without manual reconciliation? Is your state-of-the-art monitoring continuous, or annual? Is your PMS plan an operating manual, or a regulatory deliverable?
Where those questions surface gaps you'd rather close before your next surveillance audit, we'd be glad to talk. SciMed works as a long-term lifecycle partner to MedTech and IVD manufacturers, embedding the integration, governance and scientific rigour that turn a documentation programme into a sustainable, audit-ready evidence ecosystem. Download the checklist, run the internal review, and get in touch if the gaps are larger than you'd like.
Pressure-Test Your MDR Evidence Lifecycle Before Audit
The most common MDR PMS findings emerge gradually: disconnected evidence flows, stale risk conclusions, fragmented PMCF integration, and surveillance systems that no longer scale operationally.
The earlier those gaps are identified, the easier they are to remediate before they become audit findings.