Medical Device Claims Under EU MDR and UK MDR
Evidence, Intended Purpose and Compliance Risks
“A claim without evidence is not a claim. A well-supported claim is a foundation for trust, safety, and compliance.”

by Dr Sylvia Lui
Clinical Affairs Analyst, SciMed Consultancy Ltd
EXECUTIVE SUMMARY
Medical device claims are not just marketing statements - they define intended purpose and directly influence regulatory expectations under EU MDR and UK MDR.
Every claim must be supported by appropriate, traceable evidence and remain consistent across all technical, clinical and commercial documentation.
Inconsistencies or unsupported claims can lead to audit findings, compliance risks and misalignment in technical documentation.
Claims must be actively controlled throughout the product lifecycle, including post-market surveillance and PMCF activities.
Strong claims governance improves regulatory confidence, reduces risk, and supports long-term market access.
INTRODUCTION - WHY CLAIMS MATTER
Medical device claims often appear straightforward on the surface. They describe what a device does, how it performs, or what benefit it provides. However, under both the EU MDR and UK MDR frameworks, claims carry significant regulatory weight.
They are not simple statements but define the device’s intended purpose, dictating clinical evaluation requirements, risk management activities and post-market obligations. Appropriate and accurate claims are critical for determining whether the device is legally compliant and for product confidence.
For manufacturers, this means claims must be carefully controlled, consistently applied and fully supported by evidence throughout the product lifecycle. It is therefore, important to understand what a claim really is, how EU and UK regulations shape them, and why getting them right is absolutely essential for manufacturers.
STAY ALIGNED WITH EVOLVING MDR EXPECTATIONS
SciMed’s monthly newsletter, MedTech Horizon, provides practical insight on clinical evidence, claims alignment, and lifecycle compliance based on what regulators and Notified Bodies are actively challenging.
If you're responsible for maintaining compliance across technical and commercial documentation, this is designed for your role.
INDUSTRY CONTEXT & BACKGROUND
The concept of “claims” has become increasingly important under modern medical device regulation. Under previous regulatory frameworks, claims were often treated as primarily marketing statements. However, under the EU MDR (Regulation (EU) 2017/745) and the UK MDR 2002 (as amended), claims now sit much closer to the core regulatory definition of the device itself. This shift reflects a broader regulatory focus on transparency, evidence-based performance and lifecycle control of medical devices.
Claims are now routinely assessed during conformity assessment, technical documentation reviews and post-market surveillance activities. They are no longer viewed in isolation, but as part of a wider system that includes clinical evidence, risk management and real-world performance data.
As a result, manufacturers are expected to maintain clear alignment between claims, intended purpose and supporting evidence across all documentation and communication channels.
WHAT EXACTLY IS A CLAIM?
A medical device claim is any statement about a device’s performance, function, or benefit. While this may sound simple, in practice it includes anything from specific measurable outcomes (for example, “reduces complication rates”) and broader functional descriptions (for example, “designed to support recovery”).
Claims are not limited to technical documentation such as Instructions for Use. They can appear across websites, marketing materials, technical documentation, clinical evaluation reports, distributor content and even social media. If it communicates something about what the device does, it may be considered a claim. Because of this, claims must be managed consistently across both regulatory and commercial functions.
Considering these obligations, making and justifying claims become more nuanced but there clear principles under EU and UK regulations

A claim without supporting evidence is not considered valid in regulatory terms and may represent a compliance risk.
CLAIMS UNDER THE EU MDR
Under the EU MDR, although claims are not formally defined, Article 7 clearly establishes restrictions on misleading information. Manufacturers must ensure that device presentation does not mislead users regarding intended purpose, safety or performance. This applies to both explicit statements and implied meaning. This often means ensuring even subtle phrasing doesn’t overstate the device’s capabilities.
In practice, claims must not:
Attribute functions, performance or clinical effects that the device does not possess
Omit or minimise relevant risks
Suggest uses beyond the defined intended purpose
Key expectations include:
Claims must be supported by appropriate evidence
Claims must align with intended purpose
Claims must not imply unverified benefits or functions
Each claim must be traceable to relevant GSPRs
A Claims-Evidence Matrix is often used to demonstrate alignment to the GSPRs, proving the device achieves the exact performance metrics as claimed and in addition, linking individual claims to supporting data and regulatory requirements in a structured way. These matrices are helpful during a clinical evaluation as they make the relationship between wording, evidence, and legal obligations very visible.

CLAIMS UNDER THE UK MDR
Under the UK MDR, claims are governed through the intended purpose and Essential Requirements framework rather than a dedicated “claims” definition. However, the expectations remain broadly consistent with the EU MDR.
Claims must:
Strictly align with intended purpose
Be verifiable with citable evidence
Have evidence that is specific and relevant or be justifiable where appropriate
Remain consistent across all product documentation and communication channels
A key difference is that UK guidance may allow more flexibility in how evidence is structured, but not in the level of standard required. Claims still require robust justification, whether based on clinical data, analytical testing, usability studies or literature evidence.
KEY CONSIDERATIONS FOR AUDITORS & ASSESSORS
During regulatory review, claims are often assessed as part of wider technical documentation scrutiny and key focus areas typically include:
Traceability and consistency
The device must have the level of performance claimed. Claims must be consistent across IFUs, labelling, clinical documentation, marketing materials and digital platforms. Any inconsistency may raise questions about control and governance.
Alignment with intended purpose
Claims must be truthful and remain within the defined intended purpose. They must not introduce ambiguity or unintended scope expansion and they cannot not hide any real possible risks.
Specificity and measurability
Claims should be supported by robust evidence that is appropriate, measurable and proportionate to the device class and risk profile.
Risk and benefit balance
Where claims relate to clinical benefit, they must be considered in the context of device risks, supported by an appropriate benefit-risk evaluation and maintained through PMS and PMCF activities.
CHECK WHETHER YOUR CLAIMS WOULD HOLD UP UNDER AUDIT
Misaligned or unsupported claims are one of the most common causes of audit findings — particularly where intended purpose, CER evidence, and IFU language drift apart.
Use our MDR Clinical Evaluation Audit-Readiness Checklist to assess whether your claims, evidence, and documentation are aligned and defensible.
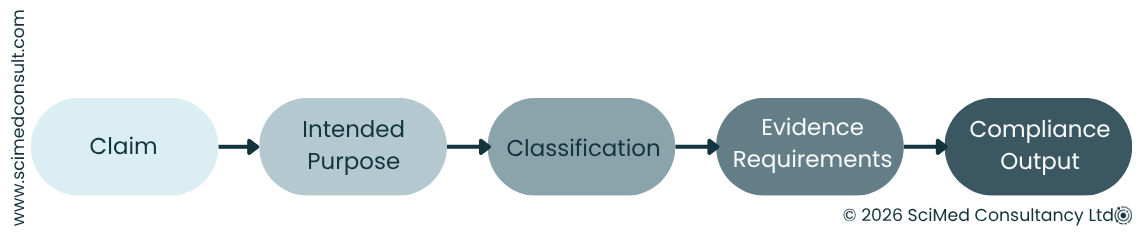
CONTINUOUS MONITORING IN THE POST-MARKET PHASE
Making claims do not remain fixed after market approval. Under both EU and UK MDR frameworks, they must be reviewed and continuously monitored as part of ongoing post-market surveillance (PMS) and, where necessary, Post‑Market Clinical Follow‑up (PMCF).
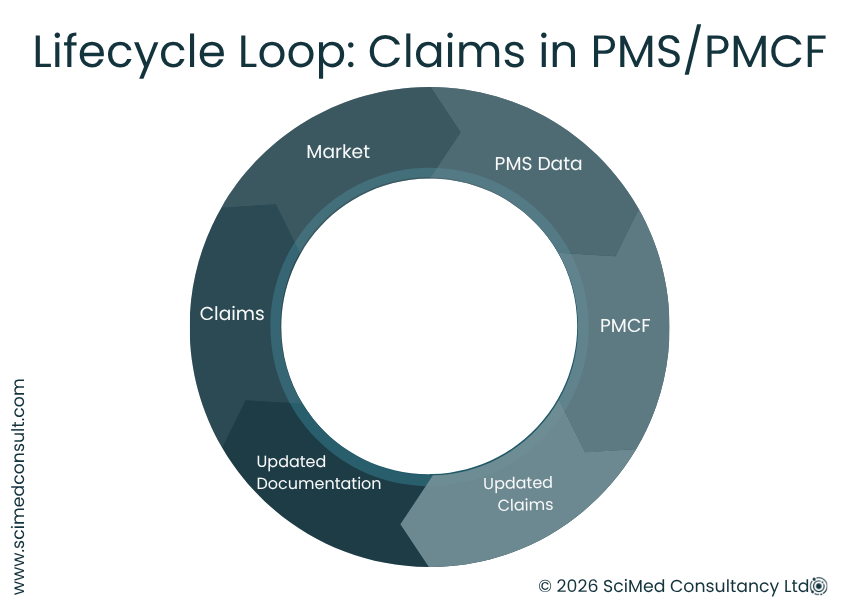
Real-world data from PMS, PMCF, complaints and vigilance reporting may confirm or challenge existing claims. Where necessary, claims may need to be updated or revised to reflect current evidence. This is particularly relevant for legacy devices or products supported by limited pre-market clinical data. The PMS data supports the proactive monitoring approach where by claims are treated as part of a continuous lifecycle process rather than a static approval milestone.
TYPES OF EVIDENCE SUPPORTING CLAIMS
The evidence required to support claims depends on the device type, classification, novelty and risk profile. Additional regulations around specific evidence requirements include:
Clinical and non-clinical evidence
Clinical evidence may include clinical investigations, PMCF studies or published literature. Non-clinical evidence may include bench testing, usability studies, analytical performance or software validation. The balance depends on the nature of the claim and device risk class. For example, non-clinical data may be used to demonstrate conformity in lower risk classes (Class IIa and below) or in specific circumstances, acceptable justification is required. Proportionate evidence with more flexible approaches for legacy and well-established devices have been recently been proposed under UK and EU law, but these changes are not yet fully implemented and are expected toward 2027.
Equivalence-based evidence
Where equivalence is being claimed, strong justification is required. Technical, biological and clinical comparability must be demonstrated, and access to equivalent device data must be sufficient, particularly under EU MDR requirements. For example, high-risk (Class III/implantable) devices, are required to have a contractual agreement of full and ongoing access to technical documentation.
Novel or innovative claims
Where claims relate to innovation or new intended use, more targeted clinical and preclinical justification is expected, alongside robust risk assessment.
CONCLUSIONS
Medical device claims play a central role in defining how a device is interpreted under both EU MDR and UK MDR frameworks. They are defined by the intended purpose, clinical evidence requirements and hence, regulatory expectations.
For manufacturers, claims must be accurate, consistent and fully supported by evidence. They must also remain aligned across all documentation and communication channels throughout the product lifecycle.
A claim without evidence is not a regulatory claim - it is a liability.
When properly controlled, claims support compliance, improve regulatory confidence and strengthen trust in the device. When poorly managed, they can introduce avoidable risk across multiple areas of regulatory and commercial activity.
Ultimately, effective claims governance is not optional - it is an essential part of maintaining medical device compliance.
USEFUL REFERENCES
EU MDR 2017/745 — Regulation (EU) 2017/745 of the European Parliament and of the Council on medical devices.
UK MDR 2002 — UK Medical Devices Regulations 2002 (SI 2002 No. 618), as amended.
European Commission — Guidance and implementation resources for MDR, including interpretation of Article 7 and GSPRs.
MHRA — UK regulatory guidance on medical devices, including expectations for claims, intended purpose, and supporting evidence.
NOT SURE IF YOUR CLAIMS ARE FULLY DEFENSIBLE?
Many teams only identify claim misalignment during audit — when inconsistencies between intended purpose, clinical evidence, and marketing materials are challenged.
A focused review can quickly clarify:
Whether your claims are appropriately supported by evidence
Where inconsistencies exist across CER, IFU, and external materials
How claims map to GSPRs and benefit–risk justification