Proving Safety Without Clinical Investigations
The role of Clinical Evaluation in Regulatory Strategy
“Clinical evaluation does not always necessitate conducting new clinical trials.”

by Dr
Nadhim
Bayatti
Senior Analyst, SciMed Consultancy Ltd
EXECUTIVE SUMMARY
Clinical evaluation under EU MDR does not always require new clinical trials; alternative evidence pathways are often acceptable for low-risk or well-established devices.
Article 61(10) provides exemptions for devices with sufficient existing clinical evidence, but careful justification and a Clinical Evaluation Report (CER) are still required.
Equivalence to an already marketed device can support compliance, but MDR imposes stricter requirements than MDD.
Post-Market Clinical Follow-up (PMCF) is mandatory and should be integrated into the clinical evaluation life cycle to strengthen evidence and support ongoing compliance.
Well-Established Technologies (WET) and “standard of care” devices can leverage literature, registry data, and post-market surveillance to build a defensible CER.
INTRODUCTION
Clinical evaluation is a cornerstone of demonstrating device safety and performance under the EU MDR, yet many manufacturers continue to assume that new clinical trials are always required. For Regulatory Affairs and R&D leaders, understanding when alternative evidence pathways are acceptable is essential for both regulatory compliance and efficient resource management. This article clarifies the regulatory framework, highlights key MDR provisions such as Article 61(10), WET and equivalence and provides practical guidance for developing a strategy for assessing and collecting the necessary clinical evidence that can be integrated in your Clinical Evaluation Report (CER) in order to satisfy Notified Body expectations.
INDUSTRY CONTEXT AND BACKGROUND
Understanding the evolution from the Medical Device Directive (MDD) to the European Union Medical Device Regulation (EU MDR) is essential for effective clinical evaluation. Under the MDD, requirements for clinical evidence were often less prescriptive, and equivalence pathways were comparatively straightforward. The MDR has introduced stricter expectations, particularly for high-risk and implantable devices, emphasizing robust documentation, continuous post-market surveillance, and lifecycle-based clinical evaluation.
For Regulatory Affairs and R&D leaders, this shift means a deeper understanding of how clinical evidence is defined, collected, and maintained is critical to achieving compliance and satisfying Notified Body expectations. Recognizing this regulatory evolution allows manufacturers to anticipate scrutiny, optimize clinical evaluation strategies, and make informed decisions on whether new clinical investigations are truly required or if alternative evidence pathways - such as equivalence or literature-based data - can be appropriately leveraged.
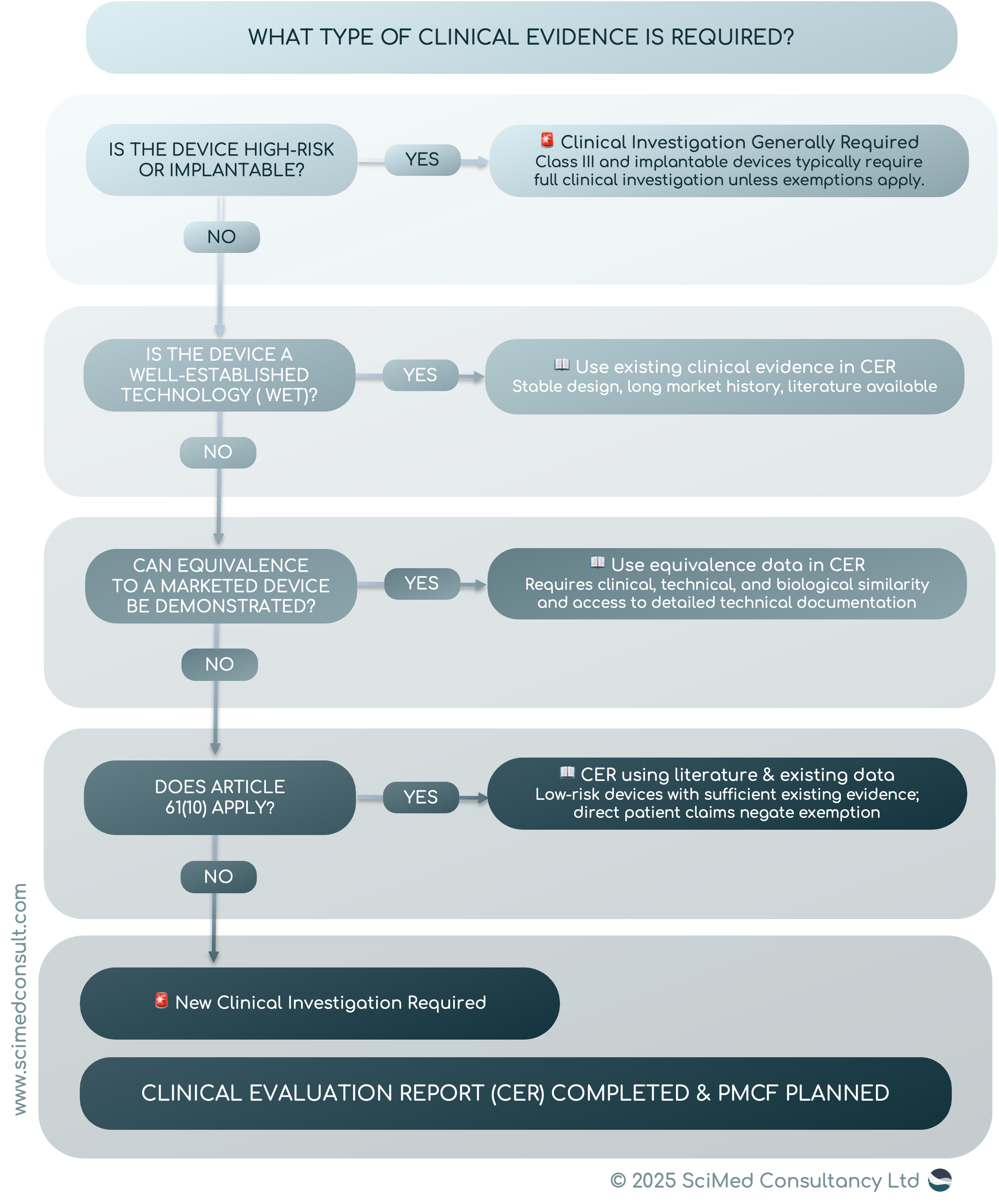
CLINICAL EVIDENCE UNDER EU MDR
The EU MDR defines ‘clinical evidence’ as clinical data and clinical evaluation results pertaining to a device of a sufficient amount and quality to allow a qualified assessment of whether the device is safe and achieves the intended clinical benefit(s), when used as intended by the manufacturer; (Article 1 of the MDR). This definition underscores two critical aspects:
Quality and quantity of data: Evidence must be scientifically sound and comprehensive.
Device-specific assessment: The evaluation must relate directly to the device or an equivalent product.
Clinical evaluation is documented in the Clinical Evaluation Report (CER), which synthesises data from clinical investigations, scientific literature, and post-market surveillance. The depth of this evaluation depends on factors such as risk class, novelty, and intended use.
RISK CLASS AND ITS IMPACT ON CLINICAL EVIDENCE REQUIREMENTS
The MDR classifies devices into four risk classes: Class I, IIa, IIb, and III, with Class III and implantable devices representing the highest risk. The higher the risk, the greater the expectation for clinical evidence. For Class III and implantable devices, clinical investigations are generally mandatory unless exemptions apply.
Conversely, low-risk devices (e.g., Class I) often rely on literature reviews and post-market data to demonstrate safety and performance. This approach is particularly relevant for well-established technologies (WET) - devices with a long history of safe use and minimal design changes.
ARTICLE 61(10): EXEMPTIONS FOR CERTAIN DEVICES
Article 61(10) of the MDR provides a critical exemption: clinical investigations may not be required for devices that are well-established and for which sufficient clinical evidence already exists. These tend to be devices that do not have ‘direct’ interaction the human body, although interaction is not excluded. However, if a manufacturer of a medical device is making clinical claims from its clinical performance that leads to a clinical benefit, then clinical data is required, and Article 61(10) no longer applies. Consideration must be given to duration, design, risks, novelty and their role in performance of the overall medical procedure. Low risk devices, without direct interaction, with no direct patient benefit and claims are ideal candidates for using article 61(10) to prove conformity to the GSPRs of the MDR. It is important to note that even with this route, a clinical evaluation must include CER, and that literature searches for the State of the Art and to identify previously unknown data need to take place.
CAN YOUR DEVICE BE SUPPORTED WITHOUT CLINICAL TRIALS?
Many manufacturers pursue Article 61(10), equivalence, or WET pathways, but struggle to demonstrate that their clinical evidence is sufficient under MDR expectations.
Our Clinical Evaluation Audit Checklist helps you assess whether your current strategy will hold up under notified body review.
It helps you identify:
Whether your justification for avoiding clinical investigations is sufficiently robust
Gaps in clinical evidence across literature, PMS, and PMCF
Weak or unsupported equivalence claims
Misalignment between your CEP, CER, and post-market strategy
Where your approach may be challenged during submission
Pro Tip
Whenever relying on equivalence or Article 61(10) exemptions, maintain clear, well-structured CER documentation. Explicitly justify why new trials are not needed to satisfy Notified Body expectations.
EQUIVALENCE: A STRATEGIC PATHWAY
Another important aspect of MDR compliance is the concept of equivalence. If a manufacturer can prove that their device is equivalent to an already marketed device with sufficient clinical evidence, they may avoid conducting new trials. Equivalence must be established across three dimensions:
Clinical: Same intended purpose and similar clinical performance.
Technical: Comparable design, materials, and specifications.
Biological: Similar tissue and body contact characteristics.
However, MDR imposes stricter requirements on equivalence compared to the previous Medical Device Directive (MDD). Manufacturers must have access to detailed technical documentation of the equivalent device - often challenging unless both devices are under the same legal manufacturer.
CHALLENGES FOR HIGH-RISK AND IMPLANTABLE DEVICES
For high-risk and implantable devices, exemptions and equivalence pathways are limited. These devices typically require clinical investigations due to their potential impact on patient safety. Even when leveraging existing data, manufacturers must provide compelling evidence that the device performs as intended under real-world conditions.
WELL-ESTABLISHED TECHNOLOGIES (WET): A PRACTICAL ADVANTAGE
WET are a defined group of devices specifically mentioned within the MDR. A specified list in Article 61(6) includes some implantable class III devices however, the term WET is not restricted to these specific devices and Article 61(8) states that additional devices similar to these might be added to that list in future.
Therefore any device that shares a number of common features of the devices that are listed below can be considered WET and the level of evidence can be less for the devices:
relatively simple, common and stable designs with little evolution
their generic device group has well-known safety and has not been associated with safety issues in the past
well-known clinical performance characteristics and their generic device group are “standard of care” devices where there is little evolution in indications and the state of the art
a long history on the market
Devices classified as WET or “standard of care” benefit from previous long-term clinical use and the presence of published literature. For these products, manufacturers can build a strong clinical evaluation report (CER) using evidence from the generic device group (benchmark or similar devices) and not restricted to the device under evaluation.
SOURCES OF CLINICAL EVIDENCE WITHOUT CLINICAL TRIALS
When clinical investigations are not feasible - due to ethical, practical, or financial constraints - manufacturers can leverage alternative sources of clinical evidence from a number of sources.
These include:
Systematic Clinical Literature Reviews
Peer-reviewed studies, meta-analyses, and clinical guidelines. This type of evidence is expected in any clinical evaluation.
Post-Market Surveillance (PMS) Data
Complaint records, vigilance reports, and trend analyses. Another important source of clinical evidence for clinical evaluation that document real world use of the device
Registry Data
National or international registries tracking device outcomes. Only applicable when such registries exist.
Equivalence Data
Technical and clinical comparison with an already marketed device.

PMCF PLANNING: BUILDING A ROBUST POST-MARKET STRATEGY
Article 61(11) of the MDR requires that the clinical evaluation and associated documentation be updated throughout the entire life cycle of the medical device based on clinical data. Thus Post-Market Clinical Follow-up (PMCF) is set out in Annex XIV part B and is not optional - and most PMCF studies take place within the intended use and without additional burdensome and invasive investigations. A strong PMCF plan should include:
Objectives
Define what gaps in clinical evidence need to be addressed (e.g., long-term safety, rare adverse events).
Methods
A number of methods for collecting evidence exist that fall short of a full blown clinical trial:
Healthcare Professional (HCP) Surveys: Collect feedback on usability, performance, and complications.
Patient Questionnaires: Gather insights on comfort, satisfaction, and clinical outcomes.
Registry Participation: Contribute to national or international device registries for longitudinal data.
Observational Studies: Monitor device performance in routine clinical practice without intervention.
Analysis of Complaints and Vigilance Reports: Identify trends and corrective actions.
Timelines
Align the plan with risk class and device complexity; higher-risk devices require more frequent updates.
Data Analysis and Reporting
Ensure findings feed into the Clinical Evaluation Report and Periodic Safety Update Reports (PSUR).

CONCLUSION
Carrying out a clinical evaluation without clinical trials depends on a number of factors, including the risk class of the device, whether it is WET, equivalence with another device is claimed, or exemption through Article 61(10) applies. Justification must be made appropriately, and in most cases a post market follow-up must be planned. This PMCF planning is not just a compliance exercise - it strengthens the manufacturer’s evidence base, supports continuous improvement, and builds trust with regulators and healthcare providers.
CLINICAL EVALUATION UNDER MDR IS INCREASINGLY ABOUT STRATEGY, NOT JUST DOCUMENTATION
MedTech Horizon, our monthly newsletter, provides guidance on clinical evidence pathways, PMCF planning, and how manufacturers are navigating regulatory expectations in practice.
USEFUL REFERENCES
The EU MDR (Reg. (EU) 2017/745) can be found here.
If you are deciding between clinical investigations, equivalence, or alternative evidence pathways, you can speak with our team for a focused discussion.