How to Prepare a Clinical Evaluation Plan Aligned with MDR Annex XIV
A Stage-by-Stage Guide
“When your CEP integrates risk management, PMS, and PMCF, you show regulators a living system of control; linking evidence, benefit–risk, and safety in one coherent strategy.”

by Dr Alastair Selby
Managing Director, SciMed Consultancy Ltd
EXECUTIVE SUMMARY
The Clinical Evaluation Plan (CEP) is mandatory under MDR Annex XIV (Part A) and is a critical first step in any Clinical Evaluation.
A structured, stepwise approach ensures alignment with EU regulatory expectations and avoids gaps that could potentially trigger notified body non-conformities.
Scope, methods, and data sources must be defined upfront, directly addressing Annex XIV requirements.
Integration with risk management, PMS, and PMCF is essential; the CEP is not a stand-alone document, but rather part of a wider documentation suite.
Early planning reduces audit risk by demonstrating a scientifically sound, reproducible approach that notified bodies expect to see.
INTRODUCTION
Manufacturers navigating EU Medical Devices Regulation (MDR) quickly discover that the Clinical Evaluation Plan (CEP) is no longer a “nice-to-have;” but rather it establishes an essential framework for your entire clinical evidence strategy. Under Annex XIV Part A of the MDR, the CEP defines the methodology, scope, and sources of clinical data that will underpin the Clinical Evaluation Report (CER).
In practice, a well-prepared CEP demonstrates control, foresight, and compliance. It is a useful tool to establish confidence with your notified body that clinical evidentiary expectations have been properly considered and [hopefully!] executed.
This guide breaks down CEP preparation into six critical steps that translate Annex XIV requirements into actionable tasks. We’ve also looked at and aligned with MDCG 2020-13, as your notified body will use this template to assess your documentation at audit.
INDUSTRY CONTEXT & BACKGROUND
The shift from the Medical Devices Directive (MDD) to the MDR represents a higher burden of clinical evidence. Annex XIV Part A explicitly requires manufacturers to plan and document how they will identify, appraise, and analyse clinical data.
Unlike under the MDD, where clinical evaluation was often addressed retrospectively, the MDR mandates a proactive, structured plan, and this CEP should integrate with other regulatory processes, most notably:
Risk management (ISO 14971),
General Safety and Performance Requirements (GSPRs; Annex I) satisfaction, and
Post-Market Surveillance (PMS) and Post-Market Clinical Follow-up (PMCF) activities.
In other words: the CEP is the anchor document of your clinical evidence strategy, bridging design, risk, and post-market systems

STAGE-BY-STAGE GUIDE TO PREPARING A CLINICAL EVALUATION PLAN
Stage 1: Define the Scope and Objectives
The CEP must begin by clearly defining the device, its intended purpose, and clinical claims. According to IMDRF guidance, scope should address:
Target the full range of patient population, use environment and conditions,
Clinical indications and intended use,
Device features relevant to performance or safety, and
Any novel or high-risk characteristics.
Pro Tip
Cross-reference with your risk management documentation to ensure consistency between identified risks and planned clinical evaluation activities.
Stage 2: Identify the Regulatory and Scientific Framework
Explicitly reference the regulatory sources that govern your CEP:
MDR Annex XIV Part A - the legal basis for required CEP content,
MDCG 2020-13 - notified body assessment expectations,
IMDRF N56 (2019) - globally harmonised principles for clinical evaluation,
Other, more specific MDCG guidance written to help guide particular types of products (for example software products, or legacy products) or compliance strategies (such as leveraging clinical evidence from equivalent devices).
This ensures traceability and reassures reviewers that the CEP is rooted in authoritative sources.
IS YOUR CLINICAL EVALUATION PLAN STRUCTURED TO MEET MDR EXPECTATIONS?
A compliant CEP is not just about including the right sections — it must show a clear, defensible methodology that aligns with Annex XIV and notified body expectations.
Our Clinical Evaluation Audit Checklist helps you assess whether your current approach will hold up in practice.
It helps you review:
Alignment between CEP scope, claims, and intended purpose
Whether your data identification and appraisal methods are sufficiently defined
Integration with risk management, PMS, and PMCF
Gaps that could later impact your Clinical Evaluation Report
Where your documentation may not meet Annex XIV expectations
Stage 3: Establish Methodology for Data Identification
Annex XIV requires manufacturers to define how they will identify data sources. This should cover:
Literature search protocol: databases, keywords, inclusion/exclusion criteria,
Clinical investigation data: ongoing or planned studies,
Clinical experience data: PMS reports, registries, adverse event databases, and
Consideration of comparable devices: either via an equivalence assessment, or a less formal use of similar device data for the purposes of establishing the state-of-the-art or other benchmarks.
Pro Tip
Document the literature search strategy and justification in the CEP itself, not just in the later CER.
Stage 4: Define Appraisal and Analysis Methods
Your CEP must describe how each piece of evidence will be evaluated for:
Relevance (fit to device claims and intended use),
Quality (study design, bias, robustness), and
Clinical significance (to performance, safety and the impact on overall benefit–risk profile).
The IMDRF model illustrates the three stages:
Identification of data,
Appraisal of each dataset, and
Overall analysis leading to conclusions.
Stage 5: Address Risk Management and Benefit–Risk Assessment
The CEP should explicitly integrate with your risk management file. This includes:
How clinical data will address residual risks identified in ISO 14971 processes, and
How benefit–risk will be continually updated as new evidence emerges.
This step demonstrates to notified bodies that your CEP is not theoretical but is tied to practical risk controls.
Stage 6: Plan for Updates, PMS, and PMCF Integration
Annex XIV requires the CEP to specify:
Frequency of CER updates (dependent upon device classification).
Triggers for updates (e.g., significant safety findings, emergence of new clinical data).
Integration with PMS/PMCF plans to ensure continuous evidence generation.
Pro Tip
Define “milestones” where notified bodies may expect to review updated evidence.
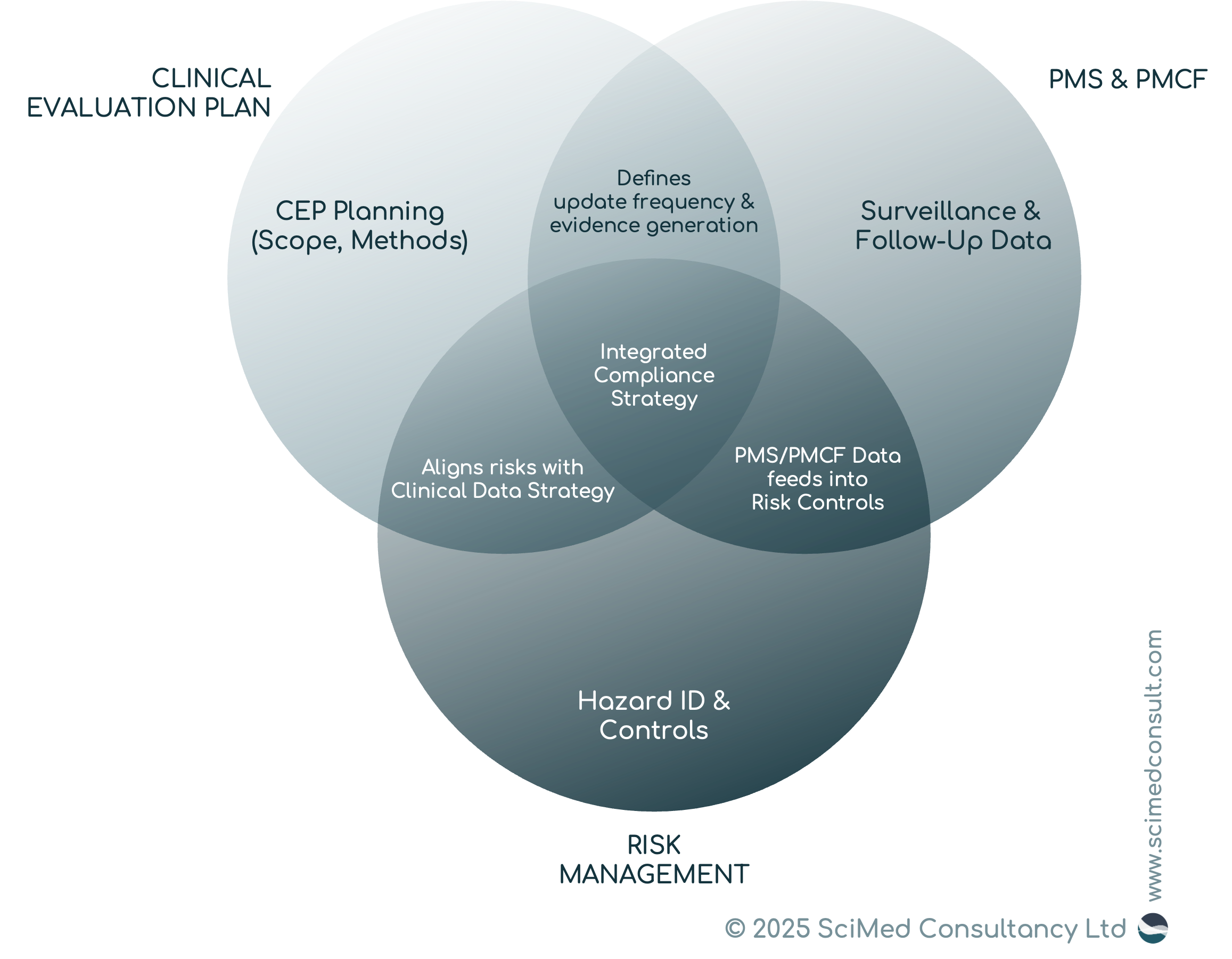
Integration of the CEP with other regulatory processes.
SUMMARY
Preparing a robust Clinical Evaluation Plan is a key foundational element of any compliant Clinical Evaluation documentation suite. By taking the time to define the scope, mapping data sources, setting appraisal methods, and considering integration with PMS, PMCF, and risk management, manufacturers create a roadmap that reassures notified bodies and reduces audit risk. A good quality CEP demonstrates foresight and scientific rigor, linking benefit–risk evaluation with real-world data. With a structured, step-by-step approach, you will not only meet regulatory requirements but also establish a sustainable compliance framework that supports product safety, performance, and long-term market confidence.
Clinical evaluation requirements under MDR continue to evolve, particularly around methodology and documentation expectations.
MedTech Horizon provides monthly guidance on CEP structure, clinical evidence strategy, and how manufacturers are aligning with notified body expectations.
If you are preparing a Clinical Evaluation Plan or reviewing your clinical evidence strategy, you can speak with our team for a focused discussion.