UK PMS Regulations vs EU MDR
What Medical Device Manufacturers Must Do from June 2025
“Have these new requirements made me panic? No; with the right updates, existing MDR-compliant systems can be efficiently adapted to meet the new MHRA requirements too.”

by Dr Nicola Edwards
Senior Analyst, SciMed Consultancy Ltd
EXECUTIVE SUMMARY
In this article, you’ll learn:
The scope and applicability of the UK post-market surveillance regulation (SI 2024/1368).
Key differences between UK PMS regulation and EU MDR post-market surveillance requirements, including timelines, reporting standards, and documentation structures.
Which devices require a PMS report vs. PSUR, and how update intervals vary based on risk classification.
What new real-world evidence and user experience data is expected under the MHRA PMS requirements.
How to adapt EU MDR-compliant systems to meet the evolving post-market surveillance requirements in Great Britain medical device regulation.
INTRODUCTION
Medical device manufacturers and IVD manufacturers must prepare for the UK’s new post-market surveillance requirements under SI 2024/1368, effective from 16June 2025. These apply to all devices sold in Great Britain, including CE-marked devices certified under the EU MDR.
This article explains:
Who is affected by the new UK PMS regulation
How UK PMS requirements differ from the EU MDR post-market surveillance framework
What documentation, timelines, and reporting formats are required
How to adapt your existing PMS system for regulatory compliance UK vs EU
Whether you're developing in vitro diagnostic devices or high-risk implantables, this guide will help align your strategy with the UK's updated MHRA post-market surveillance format.
INDUSTRY CONTEXT & BACKGROUND
The UK’s new post-market surveillance requirements for medical devices and IVDs reflect the MHRA’s post-Brexit regulatory overhaul. From June 2025, all devices sold in Great Britain must meet the new UK-specific PMS documentation and reporting expectations, even if CE-marked under the EU MDR.
While the structure aligns with the EU MDR, notable differences exist in update frequency, real-world evidence collection, and PSUR content. Manufacturers must revise their PMS plan, report templates, and classification mapping to meet MHRA PMS requirements.
WHO DO THE NEW REGULATIONS APPLY TO?
The new UK post-market surveillance rules apply to all medical device and IVD products sold in Great Britain unless:
The device is under clinical investigation, performance evaluation, or exceptional use authorization
It’s manufactured in-house by healthcare establishments
It’s a custom-made device
It has been discontinued
Devices already certified under the EU MDR must still comply with the UK PMS regulation if placed on the GB market.
HOW DO THE NEW REGULATIONS DEFINE THE PMS PERIOD?
Part 44ZC of SI 2024/1368 defines the post-market surveillance period from the first device placement or use until the last device’s operational end of life.
The UK regulations (specifically part 44ZC) define the post-market period as:
beginning with the day on which the first device of a device model is put into service by the manufacturer or placed on the market, whichever is sooner, and
ending with the end of the lifetime of the last device of that device model that is put into service by the manufacturer or placed on the market, whichever is later.

HOW DO THE UK PMS REGULATIONS COMPARE TO THOSE OF THE EU MDR?
Key differences between UK PMS regulation and EU MDR post-market surveillance include:
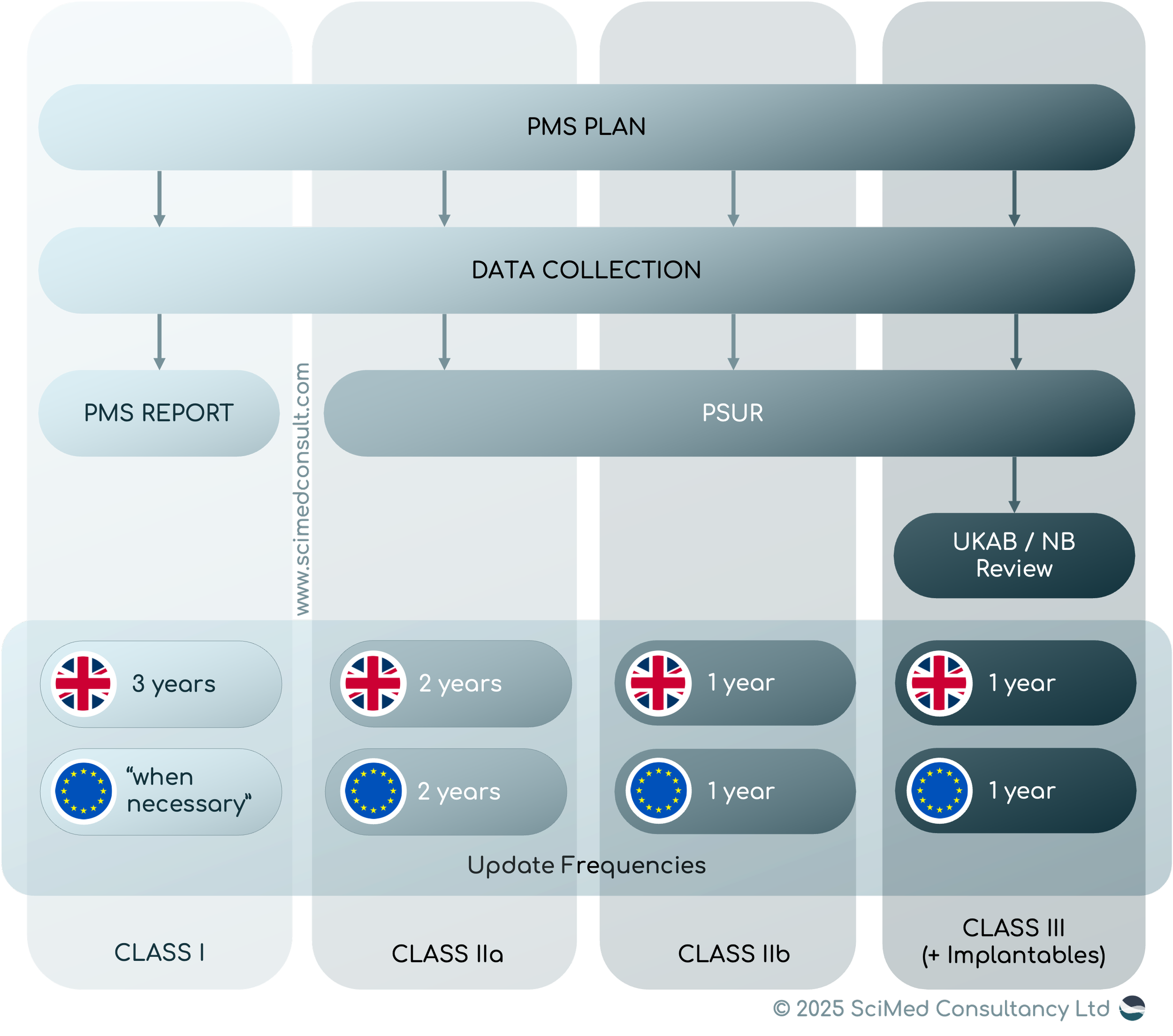
1. PMS Plan – The UK mandates inclusion of user experience data and real-world evidence through patient and public engagement. Creating a PMS plan is now compulsory for all products.
2. PMS Report – Under the EU MDR, updates are discretionary. The UK requires a PMS report:
a. Within 3 years of market placement or service
b. Updated every 3 years until the end of the PMS period
3. PSUR – The UK retains the Periodic Safety Update Report structure but adds requirements such as reporting the number of devices used without being placed on the market and estimating user population within and outside the UK.
While most EU MDR-compliant PMS documentation can transition to UK use, updates must reflect MHRA PMS requirements and include UK-specific elements.

KEEPING PACE WITH BOTH UK & EU REQUIREMENTS IS BECOMING MORE COMPLEX
Subscribe to MedTech Horizon for clear, monthly briefings on UK MHRA updates, EU MDR expectations, and how these changes affect your post-market surveillance strategy in practice.
PMSR OR PSUR – WHICH IS RIGHT FOR ME?
Under the UK PMS regulation:
A PMS report is required for class I medical devices and in vitro diagnostic devices in class A, B, or non-Annex II categories, with updates every 3 years.
A PSUR is required for class II and III medical devices and class C, D or Annex II list A/B IVDs, with minimum updates every 2 years (Class IIa) or annually (others).
A PMS plan must be created for all devices regardless of class and maintained through the full post-market surveillance period, even post-certification expiry or discontinuation.
Understanding the classification rules is only part of the challenge. Applying them correctly across your product portfolio is where most teams encounter risk.
MedTech Horizon provides practical guidance on PMS planning, PSUR expectations, and how manufacturers are adapting their systems to meet both UK and EU requirements.
IS THERE A STANDARDISED FORMAT FOR PMS DOCUMENTATION?
While an MHRA post-market surveillance format exists for PSURs (which also provides guidance on the presentation of data), there is currently no standardised format for the PMS Plan or PMS Report.
SUMMARY
The UK’s new post-market surveillance requirements align with the EU MDR but introduce specific expectations for real-world evidence, user experience data, and document frequency. To maintain compliance in Great Britain, medical device manufacturers and IVD manufacturers must review and revise their post-market clinical follow-up and post-market performance follow-up procedures accordingly. Ensuring documentation meets the MHRA PMS requirements will be critical for ongoing regulatory compliance UK vs EU in the post-Brexit landscape.
CONTINUE THE CONVERSATION
This article outlines what has changed. The next step is staying aligned as those requirements continue to evolve.
MedTech Horizon is written for MedTech and IVD teams managing post-market surveillance, clinical evaluation, and regulatory strategy across the UK and EU.
Each issue focuses on what is changing, what it means in practice, and where manufacturers need to adjust.